Make a notes on Amyloidosis pathology 2nd year MBBS
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the content needed for comprehensive notes. Let me compile everything into well-structured 2nd year MBBS pathology notes.
Amyloidosis - Pathology Notes (2nd Year MBBS)
Source: Robbins & Kumar Basic Pathology (10th ed.) and Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.)
1. DEFINITION
Amyloidosis is a group of disorders characterized by the extracellular deposition of fibrillar proteins that form insoluble aggregates, causing tissue damage and functional compromise.
- The term "amyloid" was coined because the deposits' staining resembled starch (amylose) - though the deposits are unrelated to starch.
- It is not a single disease but a group of diseases sharing the common feature of amyloid deposition.
- More than 40 different proteins can aggregate to form amyloid.
2. PHYSICAL AND CHEMICAL NATURE OF AMYLOID
Physical Nature
- By electron microscopy: continuous, nonbranching fibrils, diameter ~8-10 nm
- Each fibril consists of stacks of protofilaments arranged in a β-pleated sheet conformation
- This β-pleated sheet configuration is responsible for the characteristic Congo red staining
Chemical Composition
| Component | Percentage |
|---|---|
| Fibril proteins | ~95% |
| Serum amyloid P (SAP) component + glycoproteins | ~5% |
The Three Major Forms of Amyloid
| Type | Full Name | Source Protein | Associated Condition |
|---|---|---|---|
| AL | Amyloid Light chain | Immunoglobulin light chains (λ > κ) | Multiple myeloma, plasma cell dyscrasias |
| AA | Amyloid-Associated | Serum amyloid-associated (SAA) protein (made in liver) - 8,500 dalton protein | Chronic inflammatory diseases |
| Aβ | Beta-amyloid | Amyloid precursor protein (APP) - transmembrane glycoprotein; 4,000 dalton peptide | Alzheimer disease |
3. PATHOGENESIS
Amyloidosis results from abnormal protein folding - normally soluble proteins misfold, aggregate, and deposit as insoluble fibrils.
Two pathways of protein misfolding:
- Normal proteins that have an inherent tendency to self-associate and form fibrils, especially when produced in excess
- Mutant proteins that are prone to misfolding and aggregation
Normal quality control failure:
- Intracellular misfolded proteins are normally degraded by proteasomes
- Extracellular protein aggregates are normally taken up and degraded by macrophages
- In amyloidosis, these quality control mechanisms fail → fibrillar proteins accumulate extracellularly
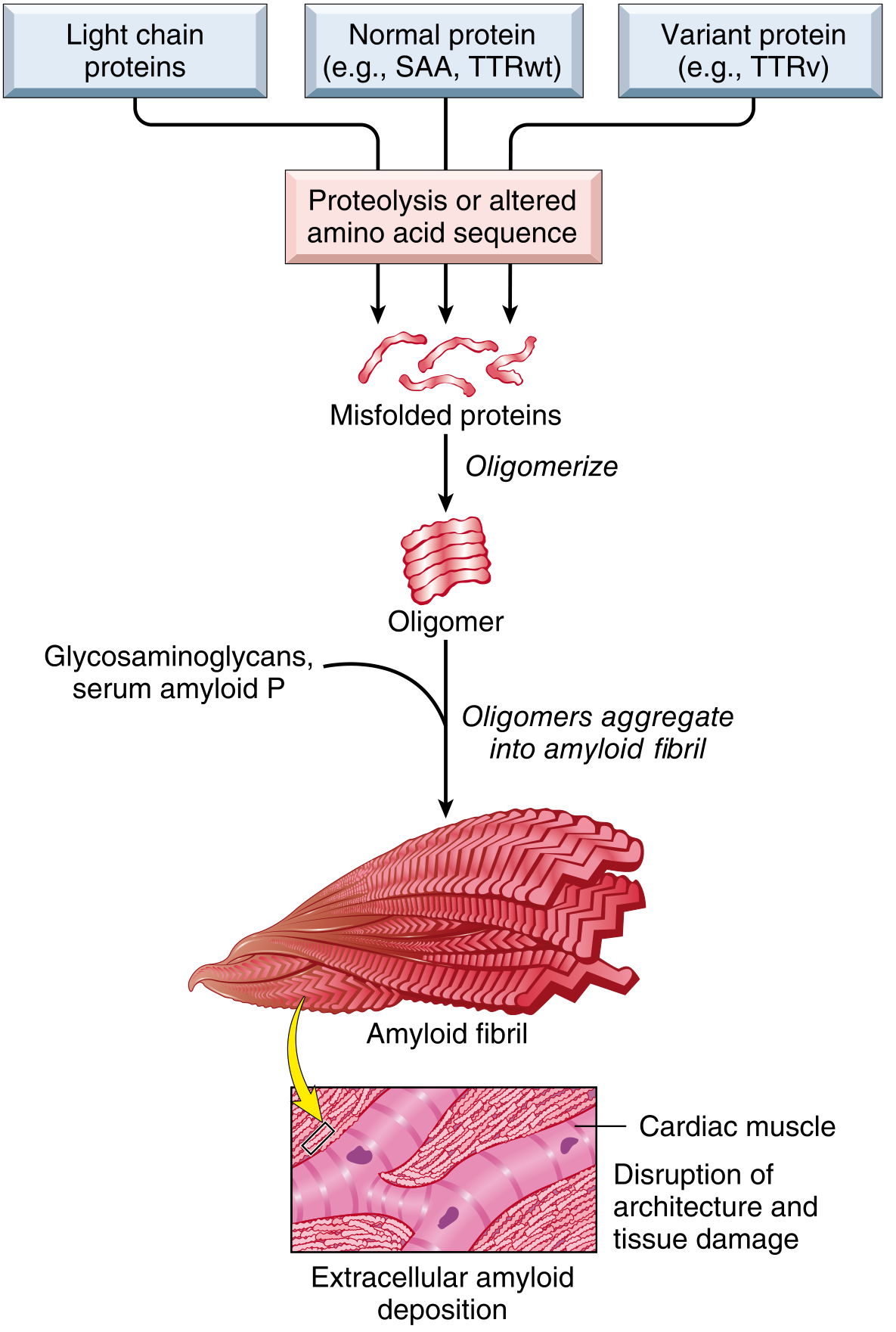
4. CLASSIFICATION
A. Systemic (Generalized) Amyloidosis
1. AL Amyloidosis (Primary Amyloidosis)
- Made of immunoglobulin light chains (complete or amino-terminal fragments), predominantly λ light chains
- Associated with: Multiple myeloma, plasma cell dyscrasias, B-cell lymphomas
- Organs most affected: Heart, GI tract, respiratory tract, peripheral nerves, skin, tongue
- This is the most common systemic amyloidosis
2. AA Amyloidosis (Secondary/Reactive Amyloidosis)
- Protein: AA protein derived by proteolysis of Serum Amyloid A (SAA) - an acute-phase protein synthesized by the liver
- SAA levels rise in chronic inflammation → sustained overproduction → amyloid deposition
- Associated conditions:
- Chronic inflammatory diseases: Rheumatoid arthritis (most common), ankylosing spondylitis, IBD
- Chronic infections: Tuberculosis, bronchiectasis, osteomyelitis
- Familial Mediterranean Fever (hereditary)
- Organs most affected: Kidneys, liver, spleen, lymph nodes, adrenals, thyroid
3. ATTR (Transthyretin) Amyloidosis
- Protein: Transthyretin (TTR) - a normal serum protein that transports thyroxine and retinol
- Two forms:
- Wild-type ATTR (senile systemic amyloidosis): Deposits in heart of elderly individuals; affects ~25% of those >80 years
- Variant ATTR (hereditary/familial): Mutant TTR deposited in peripheral nerves and heart; autosomal dominant; includes familial amyloid polyneuropathy (FAP)
- Most common form in cardiac involvement overall
4. Other Forms
- Aβ2M amyloid: β2-microglobulin in long-term dialysis patients (carpal tunnel syndrome, joint deposits)
- Aβ amyloid: In Alzheimer disease (senile plaques) and cerebral amyloid angiopathy
- AIAPP: Islet amyloid polypeptide (amylin) in type 2 diabetes mellitus - deposits in pancreatic islets
- ACal: Calcitonin-derived amyloid in medullary carcinoma of thyroid
B. Localized Amyloidosis
- Deposits in a single organ without systemic involvement
- Examples: Cutaneous amyloidosis, endocrine tumors (medullary thyroid carcinoma)
C. Hereditary/Familial Amyloidosis
- Familial Mediterranean Fever (FMF) - AA type; autosomal recessive
- Familial amyloid polyneuropathies - ATTR type (mutant transthyretin)
5. MORPHOLOGY (Gross and Histological Features)
Gross Appearance
- Affected organ is frequently enlarged
- Tissue appears gray, waxy, and firm in consistency
- Amyloid deposition begins insidiously - may not be apparent macroscopically until advanced
Histological Appearance
H&E Stain:
- Amyloid appears as an amorphous, eosinophilic, hyaline, extracellular substance
- Deposition is always extracellular, beginning between cells closely adjacent to basement membranes
- As it accumulates, it encroaches on and eventually destroys surrounding cells
- No inflammatory reaction is evoked
Congo Red Stain (Gold Standard):
- Under ordinary light: pink-red color to deposits
- Under polarized light: characteristic apple-green birefringence - the most diagnostic feature
- This reaction is shared by ALL forms of amyloid and is caused by the cross-β-pleated sheet configuration
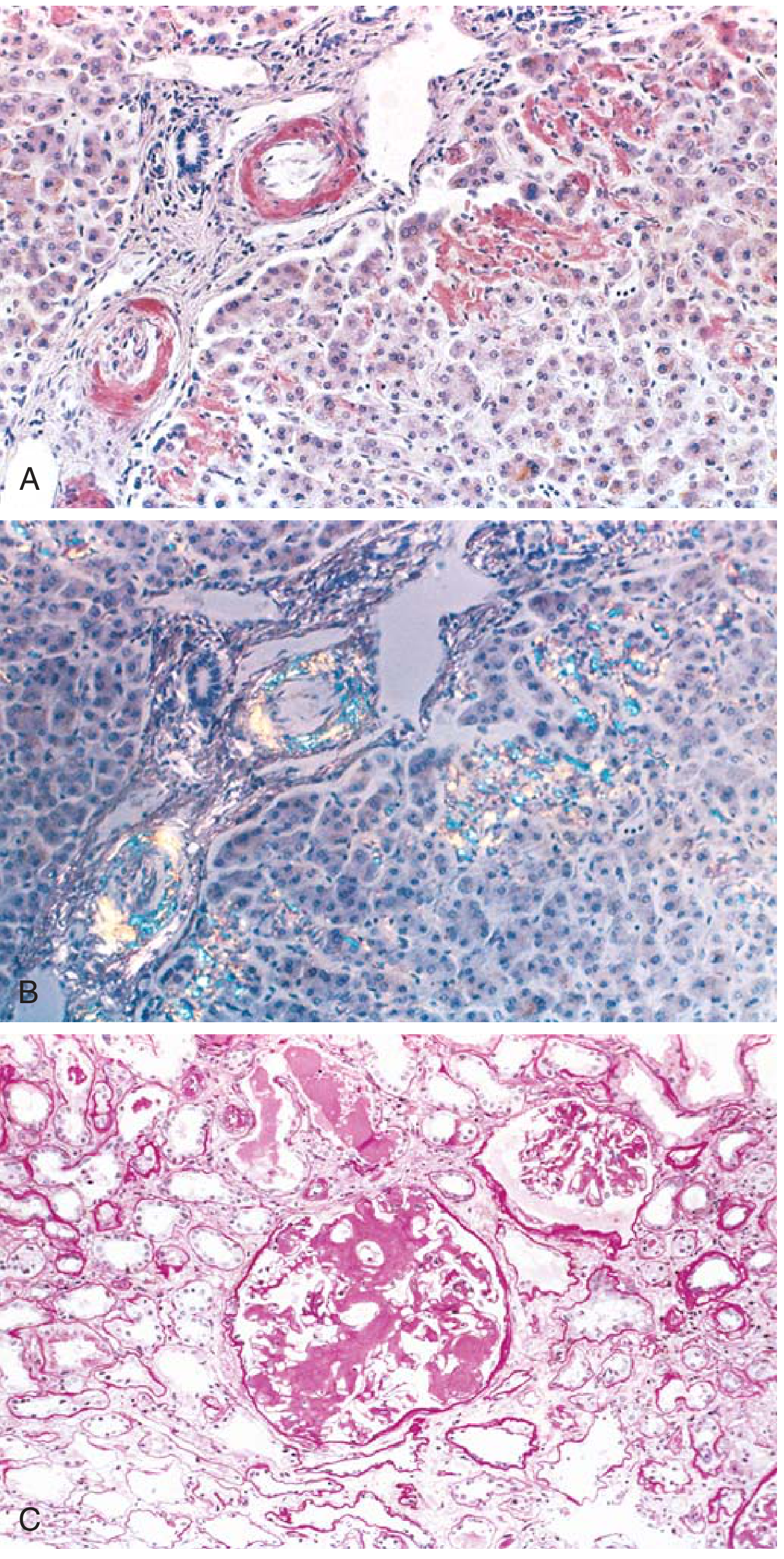
Fig: (A) Congo red stain - pink deposits in liver vessel walls and sinusoids. (B) Apple-green birefringence under polarized light - pathognomonic of amyloid. (C) Kidney - glomerular architecture almost totally obliterated by amyloid accumulation.
Additional Confirmatory Tests
- Electron microscopy: Amorphous, non-oriented, thin nonbranching fibrils (~8-10 nm)
- Immunohistochemistry: Can identify AA, AL, and TTR types specifically
- Mass spectroscopy / protein sequencing: Definitive identification of AL amyloid
6. ORGAN-SPECIFIC PATHOLOGY
Kidney (Most Common + Most Serious)
- Gross: Normal to enlarged; in advanced cases may be shrunken (ischemia from vascular narrowing)
- Histology:
- Deposits first appear in the mesangium and along glomerular basement membranes
- Causes thickening of GBM and mesangial matrix → capillary narrowing
- Progressive obliteration of capillary lumens → obsolescent glomerulus replaced by confluent masses of amyloid
- Interstitial peritubular tissue, arteries, and arterioles also affected
- Clinical: Proteinuria (nephrotic syndrome) → chronic renal failure
Spleen
- May be inapparent or cause marked splenomegaly (up to 800 g)
- Two patterns:
- Sago spleen: Deposits limited to splenic follicles → tapioca-like granules on gross inspection
- Lardaceous spleen: Deposits involve walls of splenic sinuses and red pulp connective tissue → large maplike areas
Liver
- Deposits may be inapparent or cause hepatomegaly
- Appears first in the space of Disse (between sinusoidal endothelium and hepatocytes)
- Progresses to compress and replace hepatocytes
- Rarely causes hepatic failure
Heart
- Especially in AL and ATTR amyloidosis
- Produces restrictive cardiomyopathy
- Deposits in interstitium between myofibers
- Subendocardial deposits and deposits in conducting system → arrhythmias
- Clinical: Congestive heart failure, diastolic dysfunction
Nervous System
- Peripheral neuropathy (especially in ATTR familial and AL forms)
- Autonomic neuropathy (postural hypotension, impotence, GI dysfunction)
- Carpal tunnel syndrome (AL and dialysis-associated Aβ2M)
Tongue (Macroglossia)
- Almost pathognomonic of AL amyloidosis
- Firm, rubbery enlargement of the tongue
Adrenals, Thyroid, Pituitary
- Typically in AA (secondary) amyloidosis
- May cause adrenal insufficiency
7. CLINICAL FEATURES
Depends on organs involved; onset is insidious:
| System | Manifestation |
|---|---|
| Kidney | Proteinuria, nephrotic syndrome, renal failure |
| Heart | Restrictive cardiomyopathy, CCF, arrhythmias |
| Liver | Hepatomegaly (rarely hepatic failure) |
| Spleen | Splenomegaly |
| Nervous system | Peripheral/autonomic neuropathy, carpal tunnel |
| GI tract | Malabsorption, obstruction, bleeding |
| Tongue | Macroglossia (AL type) |
| Skin | Waxy papules, periorbital purpura ("raccoon eyes" in AL) |
Important: Amyloid deposits cause tissue injury without evoking an inflammatory response and impair function by causing pressure atrophy on cells and tissues.
8. DIAGNOSIS
Tissue biopsy is required for diagnosis.
- Site of biopsy: Abdominal fat pad (most common, least invasive), rectal mucosa, gingiva, or affected organ
- Stain: Congo red - apple-green birefringence under polarized light = confirmatory
- Typing: Immunohistochemistry, mass spectroscopy, or genetic testing to determine amyloid type (essential before treatment)
9. SUMMARY TABLE
| Feature | AL (Primary) | AA (Secondary) | ATTR (Hereditary/Senile) |
|---|---|---|---|
| Protein | Immunoglobulin light chain | Serum amyloid A (SAA) | Transthyretin (TTR) |
| Associated conditions | Multiple myeloma, B-cell dyscrasia | RA, TB, chronic infections, FMF | Familial neuropathies, elderly (senile) |
| Main organs affected | Heart, tongue, nerves, GI | Kidney, liver, spleen | Heart, peripheral nerves |
| Staining | Congo red - apple-green birefringence | Same | Same |
10. KEY MNEMONICS
"AMYLOID" for causes of Secondary (AA) amyloidosis:
- A - Ankylosing spondylitis
- M - Multiple myeloma (actually causes AL, but remember as separate)
- Y - (rheumatoid)
- L - Leprosy / Lupus
- O - Osteomyelitis
- I - IBD (Crohn's)
- D - Diabetes (AIAPP in type 2)
Congo red + polarized light → Apple-green birefringence = AMYLOID (the single most important fact for exams)
11. HIGH-YIELD EXAM POINTS
- Amyloid = β-pleated sheet conformation - this is the structural basis for Congo red staining
- Congo red staining + apple-green birefringence under polarized light = gold standard diagnosis
- AL amyloidosis = associated with plasma cell dyscrasias (myeloma)
- AA amyloidosis = complication of chronic inflammatory conditions (RA, TB)
- Kidney is the most commonly and seriously affected organ
- Sago spleen = follicular deposits; Lardaceous spleen = red pulp/sinus deposits
- Macroglossia = classic sign of AL amyloidosis
- SAA protein synthesized in the liver (acute-phase reactant)
- ATTR = transthyretin; wild-type causes senile cardiac amyloidosis in elderly
- Amyloid deposits are always extracellular and cause NO inflammatory response
References: Robbins & Kumar Basic Pathology 10th ed., pp. 204-213; Robbins, Cotran & Kumar Pathologic Basis of Disease 10th ed., pp. 245-251
This is a shared conversation. Sign in to Orris to start your own chat.