Von gierke disease
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Von Gierke disease" OR "glycogen storage disease type I"
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the material needed for a thorough answer. Here it is:
Von Gierke Disease (Glycogen Storage Disease Type I)
Overview
Von Gierke disease (GSD Type I, OMIM #232200) is the most common glycogen storage disease causing hypoglycemia. It is an autosomal recessive metabolic disorder caused by deficiency of the glucose-6-phosphatase (G6Pase) system, which is essential for the final step of both glycogenolysis and gluconeogenesis - the conversion of glucose-6-phosphate (G6P) to free glucose.
- Incidence: ~1 in 100,000 live births (up to 1 in 5,420 in non-Ashkenazi North African Jews)
Subtypes
| Subtype | Defect | Gene | Chromosome | % of GSD-I |
|---|---|---|---|---|
| GSD-Ia | G6Pase hydrolase (catalytic subunit) | G6PC (G6PC1) | 17q21 | ~80% |
| GSD-Ib | G6P transporter (G6PT) | SLC37A4 | 11q23 | ~20% |
| GSD-Ic/Id | Microsomal phosphate/glucose transport | - | - | Rare |
In GSD-Ib, the G6P transporter normally moves G6P from the cytoplasm into the ER lumen where G6Pase can act on it. Loss of this transporter has the same metabolic consequence as losing G6Pase itself, plus additional immune effects.
Pathophysiology
The G6Pase system cannot hydrolyze G6P → glucose + phosphate. This blocks both glycogenolysis and gluconeogenesis from producing free glucose. The resulting metabolic cascade explains the classic tetrad:
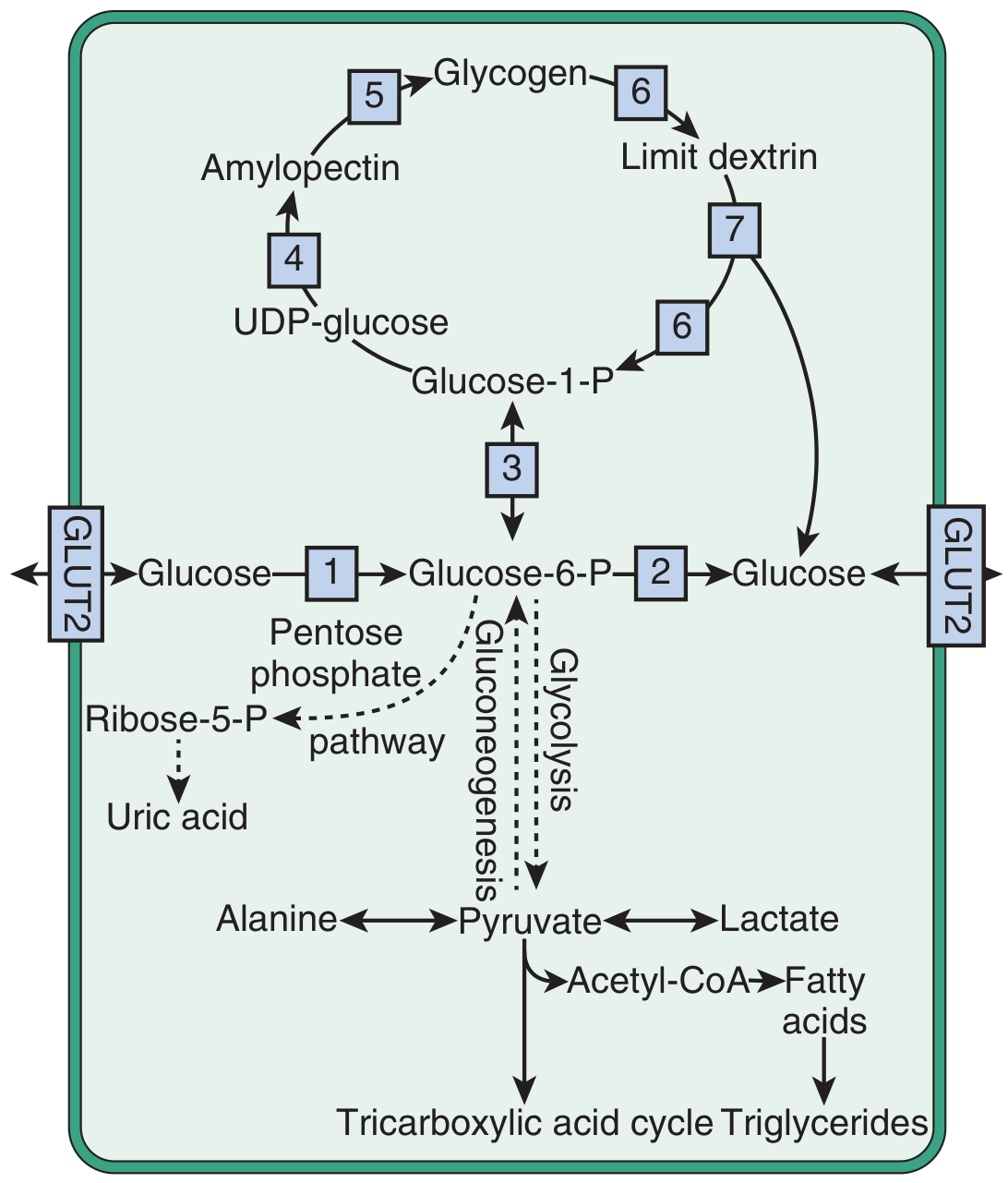
Fig. 44.4 Simplified scheme of glycogen synthesis and breakdown. Enzyme 2 = glucose-6-phosphatase - the deficient enzyme in Von Gierke disease. Accumulation of G6P drives glycolysis (→ lactic acidosis), the pentose phosphate pathway (→ hyperuricemia), and fatty acid/VLDL synthesis (→ hyperlipidemia). - Brenner & Rector's The Kidney
| Feature | Mechanism |
|---|---|
| Hypoglycemia | Impaired glycogenolysis AND gluconeogenesis - cannot release free glucose from G6P |
| Lactic acidosis | G6P accumulation drives glycolysis → excess pyruvate → lactate |
| Hyperuricemia | (1) G6P → pentose phosphate pathway → ribose-5-P → purines → uric acid; (2) lactate competes with urate for proximal tubule secretion, reducing renal urate excretion |
| Hypertriglyceridemia/hyperlipidemia | Increased VLDL and LDL synthesis; decreased lipolysis; excess acetyl-CoA → fatty acids → triglycerides |
Additionally, counter-regulatory hormones (glucagon, cortisol, catecholamines, GH) are upregulated in response to chronic hypoglycemia, further driving substrate release.
Clinical Features
Presentation in infancy:
- Hepatomegaly and/or hypoglycemic seizures with lactic acidosis in the first year of life
- Full (doll-like) cheeks, relatively thin limbs, short stature, protuberant abdomen
Classic findings:
- Hepatomegaly - massive, from glycogen and fat accumulation; no significant fibrosis
- Renomegaly - bilateral kidney enlargement
- Growth retardation
- Hypoglycemia - fasting hypoglycemia; symptoms relieved by food, worsened by exercise
- Xanthomas - from dyslipidemia (skin xanthomas)
- Macroglossia - recognized in otolaryngology literature
- Bleeding tendency - impaired platelet adhesion/aggregation; prolonged bleeding time → easy bruising, epistaxis
Later/adult complications:
- Gout (rare in children, common in adults) from sustained hyperuricemia
- Hepatic adenomas - often multiple; association with oral contraceptives and anabolic steroids; risk of malignant transformation to hepatocellular carcinoma
- Renal disease - evolves slowly; Fanconi syndrome (aminoaciduria, low-MW proteinuria, phosphaturia, bicarbonaturia), hyperfiltration, proteinuria, eventually focal segmental glomerulosclerosis and chronic kidney disease
- Pulmonary hypertension
- Steatohepatitis, pancreatic beta-cell failure
- Insulin resistance
GSD-Ib specific:
- Chronic neutropenia and neutrophil/monocyte dysfunction → recurrent bacterial infections, oral and intestinal mucosal ulceration, inflammatory bowel disease-like enterocolitis
Biochemical Profile
| Lab | Finding |
|---|---|
| Blood glucose | Low (fasting hypoglycemia) |
| Lactate | Elevated |
| Uric acid | Elevated |
| Triglycerides | Markedly elevated |
| LDL/VLDL | Elevated |
| Blood bicarbonate | Low (metabolic acidosis) |
| Glucagon stimulation test | Blood glucose rise <4 mmol/L (abnormal) |
Diagnosis
- DNA testing (first-line): Identification of pathogenic variants in G6PC1 (GSD-Ia) or SLC37A4 (GSD-Ib)
- Glucagon stimulation test (1 mg IM): abnormal response - glucose rise <4 mmol/L at 30 minutes
- Liver biopsy (rarely needed now): prominent glycogen and fat storage, large lipid vacuoles, hepatocyte distension, steatosis with little fibrosis; electron microscopy shows massive cytoplasmic glycogen displacing organelles
- Enzyme analysis of fresh/frozen liver samples can distinguish Ia from Ib
Treatment
The goal is to maintain normoglycemia to prevent the metabolic cascade.
- Uncooked cornstarch (slow-release glucose): 1.75-2.5 g/kg at bedtime maintains glucose >3.9 mmol/L for 7+ hours in adults; cornerstone of management
- Continuous nocturnal nasogastric glucose feeds (especially in young infants)
- High-carbohydrate diet throughout the day
- Allopurinol - for hyperuricemia/gout
- Lipid-lowering agents - for hypertriglyceridemia
- ACE inhibitors - for renal protection (proteinuria)
- Citrate supplementation - for renal tubular acidosis and urolithiasis prevention
Monitoring targets (European Study Group guidelines):
- Preprandial glucose >3.5-4.0 mmol/L (60-70 mg/dL)
- Urine lactate:creatinine <0.06 mmol/mmol
- Serum uric acid in high-normal range
- Venous bicarbonate >20 mmol/L
- Serum triglycerides <6.0 mmol/L (531 mg/dL)
Liver transplantation - corrects the hepatic enzyme deficiency and the metabolic derangements but does not correct the renal defect. Kidney transplantation has been performed but does not correct hypoglycemia.
Enzyme replacement therapy is not currently available for GSD-I (unlike GSD-II/Pompe disease).
Anesthetic Considerations
- High hemorrhage risk from platelet dysfunction - requires pre-op evaluation
- Avoid lactate-containing fluids (Ringer's lactate) - these patients cannot convert lactate to glycogen
- IV glucose-containing fluids mandatory when fasting perioperatively
- Frequent perioperative blood glucose monitoring
- Procedures these patients may require: liver tumor ablation/resection, liver transplant, urolithiasis surgery, renal transplant
Key Distinguishing Points (vs. Other GSDs)
| Feature | GSD I (Von Gierke) | GSD II (Pompe) | GSD III (Cori) | GSD V (McArdle) |
|---|---|---|---|---|
| Enzyme | G6Pase | Acid maltase (GAA) | Debranching enzyme | Muscle phosphorylase |
| Hypoglycemia | Severe | No | Mild | No |
| Organ | Liver, kidney | Heart, muscle, liver | Liver, muscle | Muscle |
| Lactic acidosis | Yes | No | Mild | No (↑ after exercise) |
| Hyperuricemia | Yes | No | Mild | No |
Sources: Brenner & Rector's The Kidney; Miller's Anesthesia, 10e; Goldman-Cecil Medicine; Harper's Illustrated Biochemistry, 32e
Recent evidence note: A 2026 systematic review (PMID: 41603948) addressed SGLT-2 inhibitors in GSD-Ib for managing neutropenia-associated digestive symptoms - an emerging treatment consideration specific to the Ib subtype.
This is a shared conversation. Sign in to Orris to start your own chat.