Pathophysiology of Parkinson disease
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Reading File
I have now gathered comprehensive, detailed information from multiple authoritative textbooks. Let me compose the full answer.
Pathophysiology of Parkinson Disease
1. Overview
Parkinson disease (PD) is a progressive neurodegenerative disorder defined by a hypokinetic movement disorder resulting from the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The classical motor triad - resting tremor, bradykinesia, and cogwheel rigidity - plus postural instability, reflects disruption of the cortico-striato-thalamo-cortical motor circuit. Symptom onset is typically between 55 and 65 years of age, with ~1% of individuals over 65 affected. - Robbins & Kumar Basic Pathology, p. 854
2. Core Neuropathology
2a. Substantia Nigra Degeneration
The hallmark pathological finding is the selective loss of pigmented dopaminergic neurons in the SNpc. Grossly, the substantia nigra appears pale on midbrain cross-section due to the loss of neuromelanin-containing neurons (normally the SNpc gives the substantia nigra its characteristic dark appearance). The locus ceruleus and other pigmented brainstem nuclei are also affected. - Neuroanatomy through Clinical Cases 3rd Ed., p. 791
By the time motor symptoms appear, approximately 60-80% of dopaminergic neurons have already been lost and striatal dopamine is reduced by ~80%. Aging alone contributes some cell loss, but the depletion in PD far exceeds that expected from normal aging. - Adams and Victor's Principles of Neurology, 12th Ed., p. 1089
2b. Lewy Bodies - The Pathological Signature
Remaining dopaminergic neurons contain Lewy bodies: eosinophilic, cytoplasmic, round-to-elongated inclusions with a dense pink core and faint halo on H&E staining.
Composition of Lewy bodies:
- α-synuclein (the major component) - normally a synaptic protein involved in synaptic vesicle recycling
- Ubiquitin
- Neurofilaments and other proteins
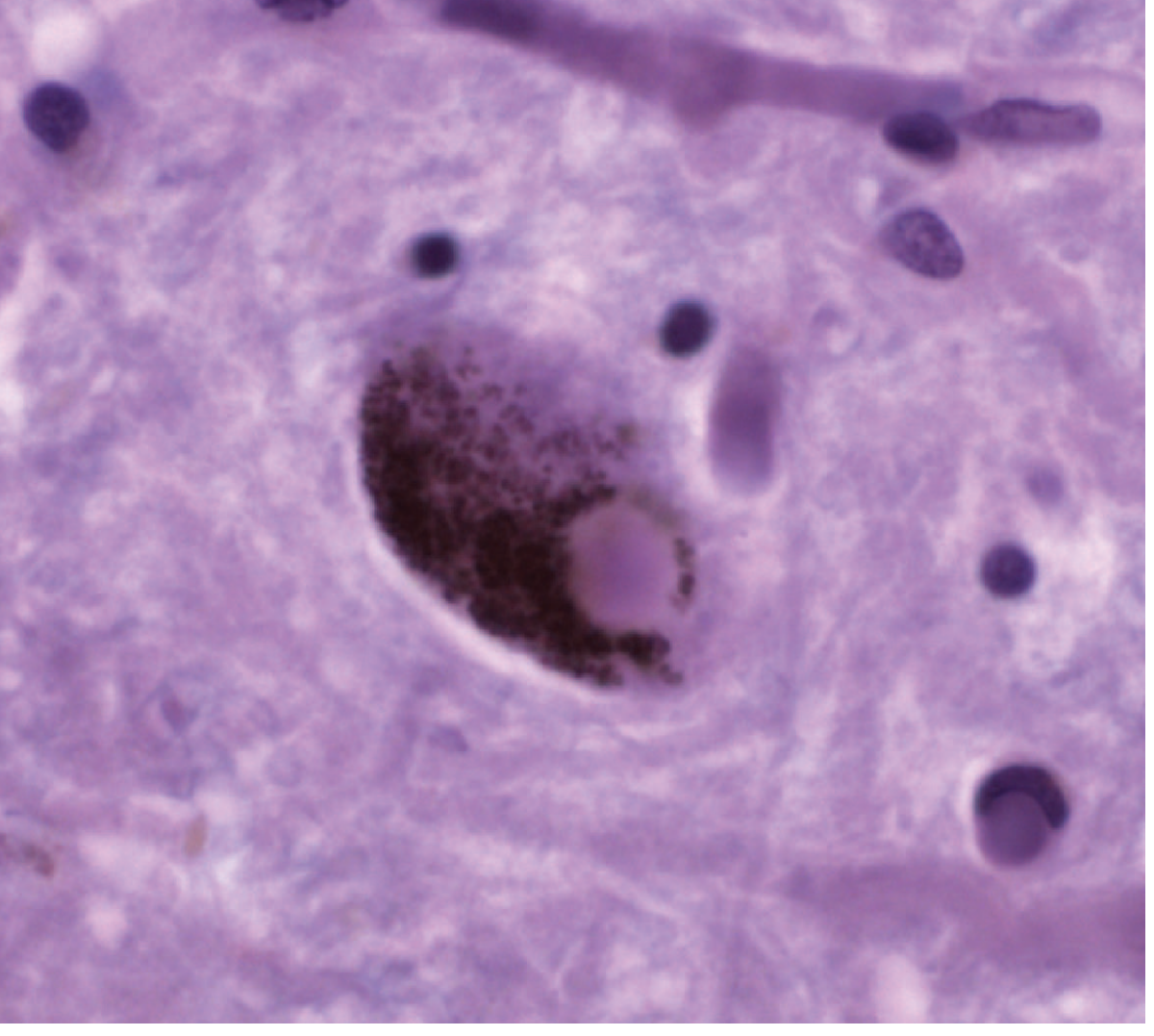
Figure: Photomicrograph of a Lewy body (round, dense cytoplasmic inclusion) in a pigmented dopaminergic neuron of the substantia nigra. - Adams and Victor's Principles of Neurology, 12th Ed.
3. Molecular Pathogenesis
3a. α-Synuclein Aggregation
α-Synuclein, normally a soluble synaptic protein, misfolds and aggregates into insoluble fibrils that form Lewy bodies. This aggregation is a central event in PD pathogenesis:
- Point mutations and gene duplications of the α-synuclein gene (SNCA) cause autosomal dominant PD in rare families
- Aggregated α-synuclein is toxic to neurons
- Evidence from the vagotomy epidemiologic studies and the Braak staging hypothesis suggests α-synuclein pathology may originate in the enteric nervous system and olfactory bulb before spreading rostrally to the midbrain via prion-like cell-to-cell propagation - Adams and Victor's, p. 1089
3b. Impaired Protein Clearance (Autophagy/Lysosomal Dysfunction)
Multiple genetic loci implicate defective autophagy and lysosomal degradation:
| Gene | Protein | Pathway | Inheritance |
|---|---|---|---|
| SNCA | α-synuclein | Synaptic transmission | AD (duplications/mutations) |
| Parkin | E3 ubiquitin ligase | Mitophagy, endosomal trafficking | AR |
| LRRK2 | Leucine-rich repeat kinase 2 | Kinase (mechanism unclear) | AD - most common cause of familial PD |
| GBA1 | Glucocerebrosidase | Lysosomal enzyme | Risk factor (heterozygous mutation) |
The GBA1 (glucocerebrosidase) connection is particularly telling - a lysosomal enzyme deficiency is a major risk factor, directly linking impaired lysosomal turnover to PD pathogenesis. - Robbins & Kumar Basic Pathology, p. 854
3c. Mitochondrial Dysfunction and Oxidative Stress
The neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) reproduces clinical and pathological parkinsonism in humans and primates:
- MAO converts MPTP to MPP+ (the active toxin)
- MPP+ is selectively taken up by dopaminergic neurons via their dopamine transporters
- MPP+ inhibits mitochondrial Complex I, blocking oxidative phosphorylation and causing cell death through energy failure and ROS production
This MPTP model strongly implicates mitochondrial dysfunction and oxidative stress in sporadic PD pathogenesis. Mutant Parkin protein also plays a role in mitophagy (clearance of damaged mitochondria). - Adams and Victor's Principles of Neurology, 12th Ed., p. 1089
3d. Braak Staging - A Progression Hypothesis
Braak and colleagues proposed that α-synuclein pathology follows a caudo-rostral staging:
- Stages 1-2: Dorsal vagal nucleus, anterior olfactory nuclei (explains early anosmia and autonomic symptoms)
- Stages 3-4: Substantia nigra and midbrain (motor symptoms emerge)
- Stages 5-6: Neocortex (dementia)
This staging explains non-motor symptoms (anosmia, constipation, REM sleep behavior disorder) that precede motor symptoms by years to decades. - Adams and Victor's Principles of Neurology, 12th Ed.
4. Circuit-Level Pathophysiology: Basal Ganglia Motor Loops
The motor symptoms of PD are a direct consequence of dopamine depletion disrupting the cortico-striato-thalamo-cortical circuit through two key pathways:
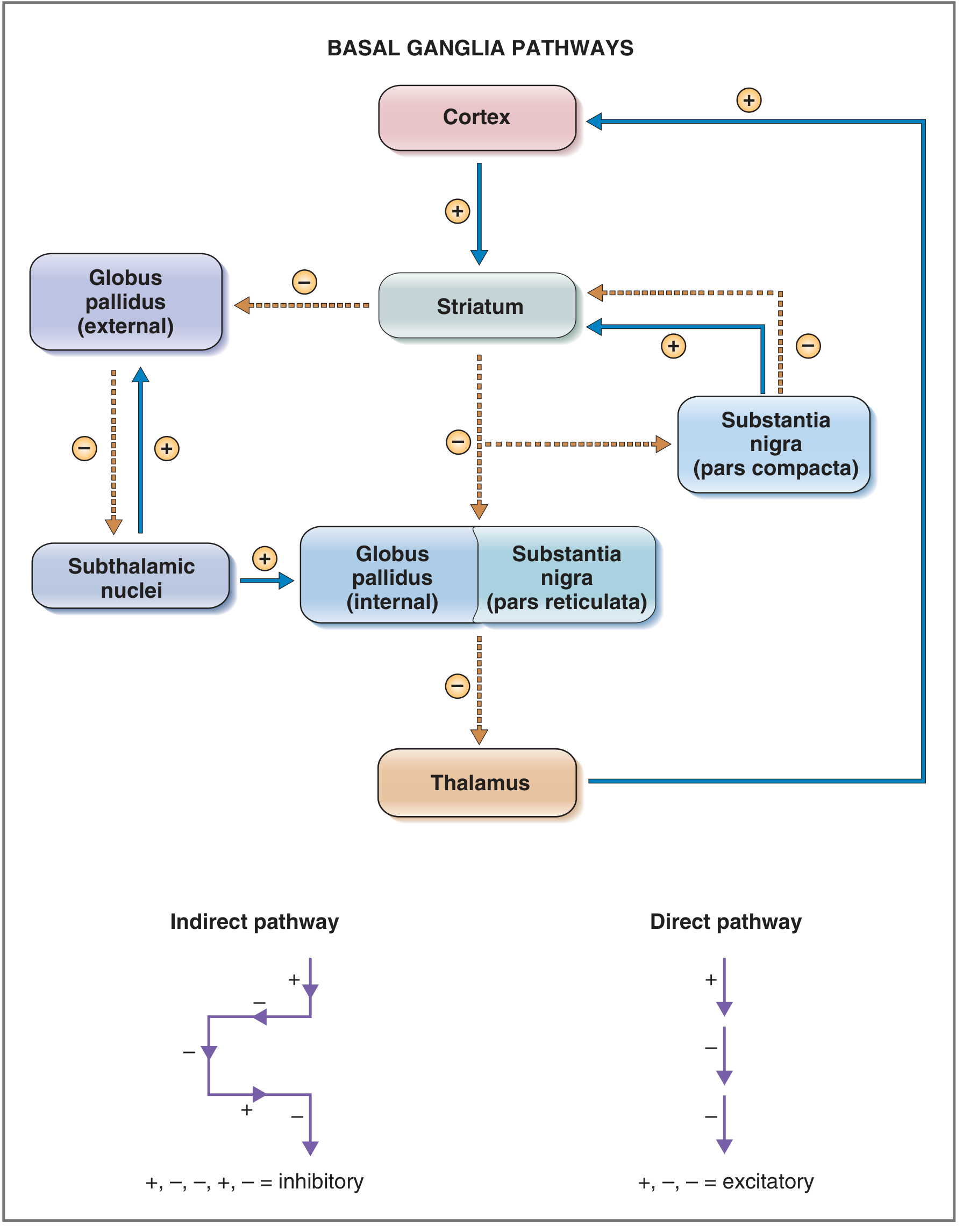
Figure: Basal ganglia direct and indirect pathways. Blue arrows = excitatory; dashed brown arrows = inhibitory. - Costanzo Physiology 7th Ed.
Normal Physiology
Direct pathway (pro-kinetic): Cortex → Striatum → GPi/SNpr (inhibited) → Thalamus (disinhibited) → Cortex (excited)
- Net result: thalamo-cortical excitation → movement facilitated
- Dopamine acts via D1 receptors to EXCITE striatal neurons of this pathway
Indirect pathway (anti-kinetic): Cortex → Striatum → GPe (inhibited) → STN (disinhibited) → GPi (excited) → Thalamus (inhibited) → Cortex (reduced)
- Net result: thalamo-cortical inhibition → unwanted movements suppressed
- Dopamine acts via D2 receptors to INHIBIT striatal neurons of this pathway
Summary: Dopamine normally excites the direct pathway AND inhibits the indirect pathway - both effects produce a net excitatory output to the thalamus and cortex, facilitating movement. - Neuroanatomy through Clinical Cases, p. 779; Adams and Victor's, p. 1089
In Parkinson Disease
Loss of SNpc dopamine causes:
- Direct pathway underactivity: Less D1 stimulation → striatum fails to inhibit GPi → GPi remains overactive → GPi over-inhibits thalamus
- Indirect pathway overactivity: Less D2 inhibition → striatum fails to inhibit GPe → GPe inhibits STN less → STN becomes overactive → STN over-excites GPi → GPi over-inhibits thalamus
Both pathways converge to produce EXCESS INHIBITION of the thalamus, reducing thalamo-cortical drive and impairing initiation and execution of voluntary movement (bradykinesia, hypokinesia). - Adams and Victor's Principles of Neurology, 12th Ed., p. 1089
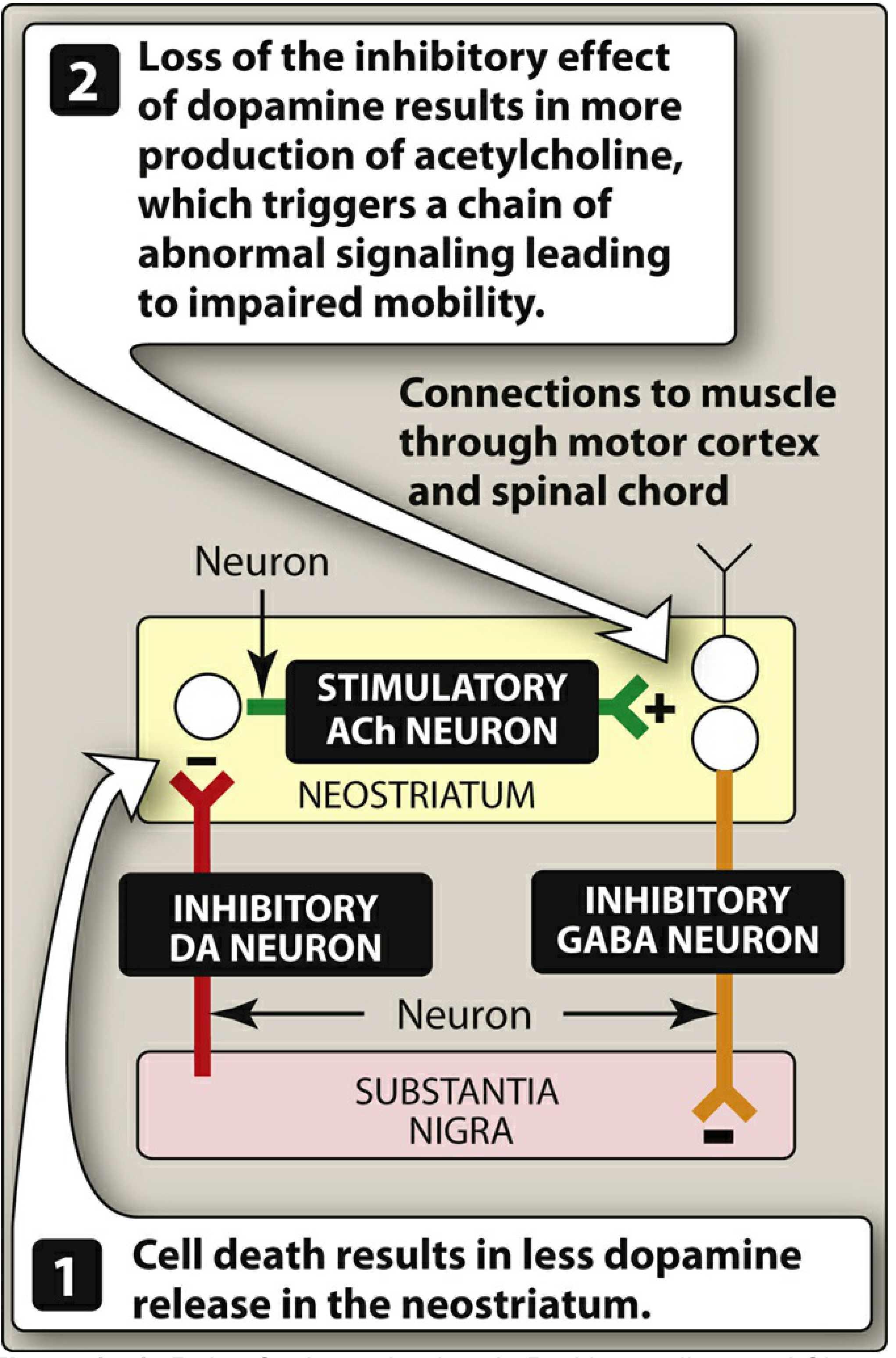
Figure: Role of the substantia nigra in PD. Dopamine cell death → loss of dopamine inhibition → relative ACh overactivity → impaired motor control. - Lippincott Illustrated Reviews: Pharmacology
The Dopamine-Acetylcholine Imbalance
The neostriatum normally maintains a balance between dopaminergic (inhibitory) and cholinergic (stimulatory) neurons. In PD:
- Loss of dopaminergic input removes inhibition of cholinergic interneurons
- Relative overactivity of acetylcholine results
- This imbalance contributes to motor symptoms and explains why anticholinergic drugs (e.g., benztropine) have therapeutic benefit in PD - Lippincott Illustrated Reviews: Pharmacology, p. 513
Oscillatory Dysfunction and Tremor
The classic basal ganglia model does not fully explain tremor. Current evidence suggests:
- Pathological β-oscillations (13-30 Hz) arise in the cortico-basal ganglia circuit and characterize bradykinesia and akinesia
- Parkinsonian tremor involves abnormal thalamocortical and cerebellar-cortical oscillations - it is modulated by the cerebellum, not purely a basal ganglia phenomenon
- Physiological γ-activity (>30 Hz) re-emerges with clinical improvement (e.g., with levodopa or DBS) - Bradley and Daroff's Neurology in Clinical Practice
5. Non-Motor Pathophysiology
Because α-synuclein pathology extends well beyond the SNpc, PD affects multiple neurotransmitter systems and circuits:
| System Affected | Non-Motor Manifestation |
|---|---|
| Enteric nervous system / dorsal vagal nucleus | Constipation, dysphagia |
| Olfactory bulb | Anosmia (often precedes motor onset by years) |
| Locus ceruleus (noradrenergic) | Sleep disturbances, depression |
| Raphe nuclei (serotonergic) | Depression, anxiety |
| Autonomic ganglia | Orthostatic hypotension, urinary dysfunction |
| Pedunculopontine nucleus | Falls, axial rigidity |
| Cerebral cortex (Stages 5-6 Braak) | Dementia (Parkinson disease dementia, overlap with Lewy body dementia) |
6. Summary Diagram of Pathophysiology
Genetic/Environmental triggers
↓
α-synuclein misfolding & aggregation
+ Mitochondrial Complex I dysfunction
+ Impaired autophagy/lysosomal clearance
↓
Death of dopaminergic neurons in SNpc
↓
┌──────────────────────────────────────┐
│ Striatal Dopamine Depletion │
└──────────────────────────────────────┘
↓ ↓
Direct pathway ↓ Indirect pathway ↑
(less D1 activation) (less D2 inhibition)
↓ ↓
GPi overactivity (increased inhibitory output)
↓
Thalamus over-inhibited
↓
Reduced thalamo-cortical excitation
↓
BRADYKINESIA / HYPOKINESIA / RIGIDITY
(Tremor involves additional thalamo-cerebellar oscillatory circuits)
Key Sources
- Robbins & Kumar Basic Pathology, p. 854 - pathogenesis, morphology, genetics
- Adams and Victor's Principles of Neurology, 12th Ed., pp. 1089-1092 - pathology, molecular mechanisms, MPTP model
- Neuroanatomy through Clinical Cases, 3rd Ed., pp. 779, 791 - circuit-level pathophysiology, direct/indirect pathways
- Costanzo Physiology, 7th Ed., p. 116 - basal ganglia circuits and disease
- Lippincott Illustrated Reviews: Pharmacology, pp. 512-514 - dopamine-ACh imbalance
- Bradley and Daroff's Neurology in Clinical Practice - oscillatory pathophysiology, non-motor features
This is a shared conversation. Sign in to Orris to start your own chat.