Bernard soulier syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Bernard-Soulier syndrome"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Here is a thorough overview of Bernard-Soulier Syndrome:
Bernard-Soulier Syndrome (BSS)
Definition & Genetics
Bernard-Soulier syndrome is an autosomal recessive inherited bleeding disorder caused by deficiency or dysfunction of the GPIb-IX-V complex on the platelet surface. Mutations occur in the genes GPIBA, GPIBB, and GP9. It is classified as a defect of platelet adhesion.
-
The GPIb-IX-V complex is the receptor for von Willebrand factor (vWF) - without it, platelets cannot bind to the subendothelial vWF at sites of vascular injury, impairing primary hemostatic plug formation.
-
A monoallelic (autosomal dominant) form exists, which is much milder and associated with large platelets and mildly low platelet counts; it can only be confirmed by flow cytometry or genetic testing.
-
Carrier state can present as Mediterranean macrothrombocytopenia, a relatively common cause of mild thrombocytopenia in southern Europe.
-
Goldman-Cecil Medicine, p. 1663
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 3019
Pathophysiology
Under normal conditions, when a vessel is injured, vWF in the subendothelial matrix is exposed. Platelet GPIbα (part of the GPIb-IX-V complex) binds this vWF, anchoring the platelet to the vessel wall. In BSS, this adhesion step fails entirely, so no primary plug can form regardless of platelet count or other function.
Clinical Features
- Mucocutaneous bleeding - the hallmark presentation (epistaxis, gingival bleeding, menorrhagia, easy bruising, petechiae, purpura)
- Bleeding typically presents early in life and can range from mild to severe
- Postpartum hemorrhage occurs in ~60% of affected pregnancies, with ~50% requiring blood transfusion
- Severity is variable; some patients have minimal symptoms while others have life-threatening bleeds
Laboratory Findings
Blood Count & Smear
- Thrombocytopenia (mild to moderate) - platelet count is low but not always severely so
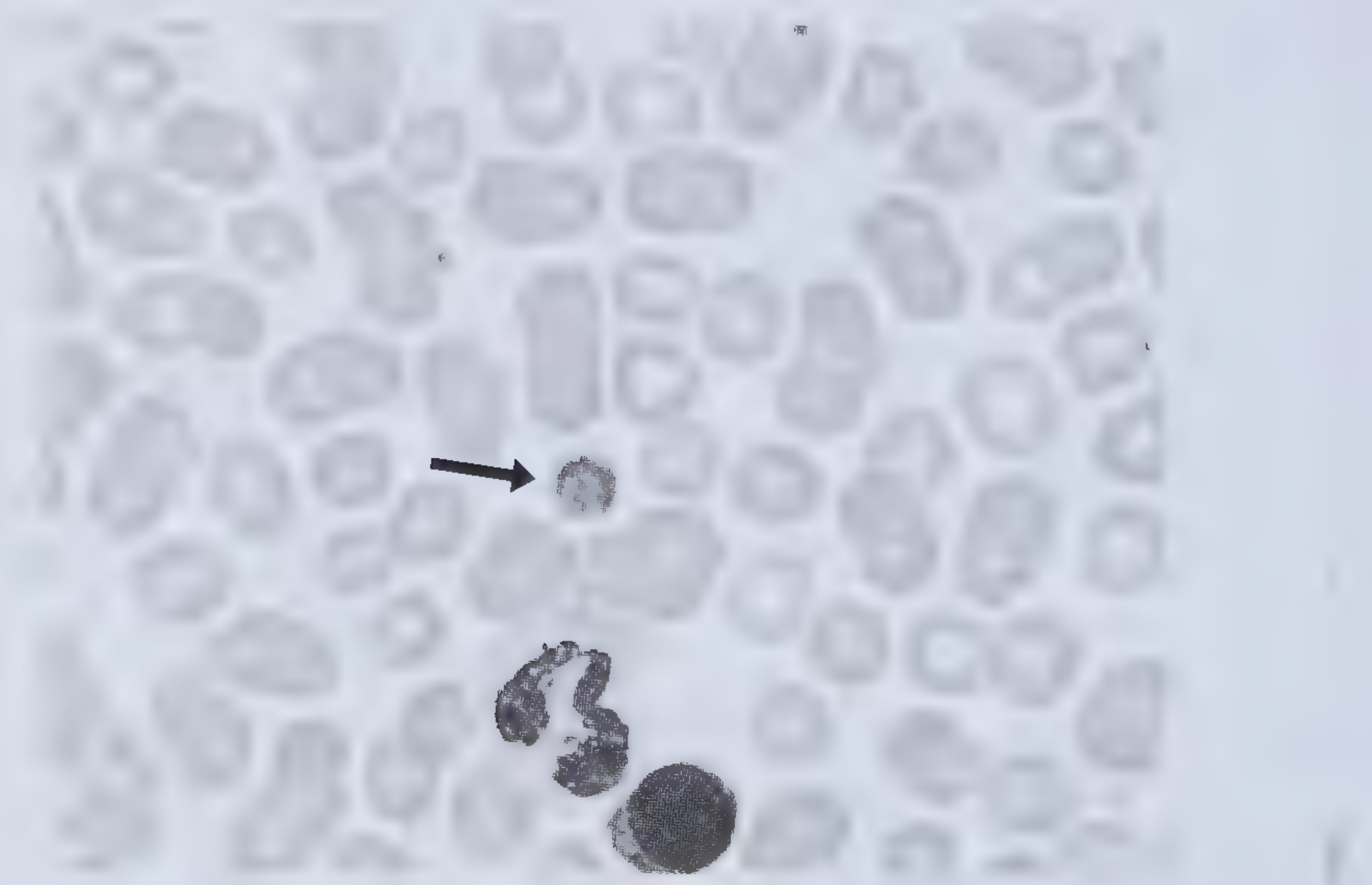
- Giant (macro) platelets on peripheral blood smear - a hallmark feature
Peripheral blood smear showing giant platelet (arrow) in BSS:
Platelet Aggregation Studies (Light Transmission Aggregometry - LTA)
| Agonist | BSS Response | Key Point |
|---|---|---|
| Ristocetin | Absent/severely reduced | Ristocetin requires GPIb-vWF interaction |
| ADP | Normal | |
| Arachidonic acid | Normal | |
| Collagen | Normal | |
| Epinephrine | Normal |
This pattern (fail ristocetin only, respond to all others) is shared with vWD but can be distinguished:
-
Unlike vWD: BSS peripheral smear shows large platelets
-
Unlike vWD: BSS fails to aggregate even when normal plasma is added (because the defect is in the platelet receptor, not the plasma vWF)
-
Quick Compendium of Clinical Pathology 5th ed., p. 462-481
Coagulation Tests
- PT and aPTT: normal
- Bleeding time / PFA-100: prolonged
Flow Cytometry
Confirmatory test - shows absent or severely reduced surface expression of GPIbα on platelets, with normal GPIIb-IIIa expression.
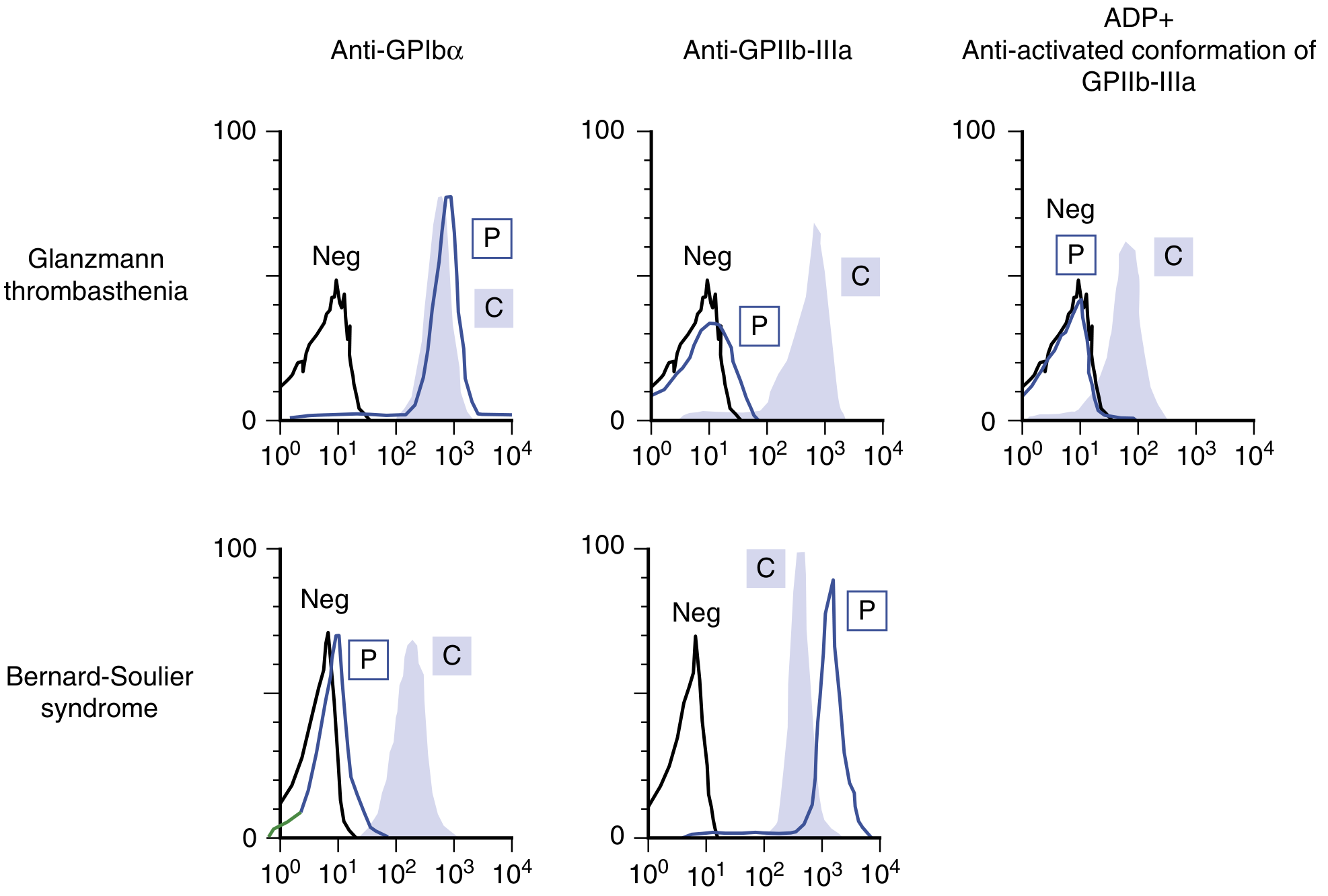
Flow cytometry: In BSS (lower row), the patient shows near-absent GPIbα (left column, P peak at low fluorescence) while GPIIb-IIIa is normal. This is the opposite pattern from Glanzmann thrombasthenia (upper row).
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 1932
Comparison with Glanzmann Thrombasthenia
| Feature | Bernard-Soulier | Glanzmann Thrombasthenia |
|---|---|---|
| Deficient receptor | GPIb-IX-V | GPIIb-IIIa |
| Receptor's ligand | vWF | Fibrinogen |
| Defect type | Adhesion | Aggregation |
| Platelet size | Giant | Normal to small |
| Ristocetin response | Absent | Normal |
| All other agonists | Normal | Absent |
| Clot retraction | Normal | Abnormal |
| Inheritance | AR | AR |
Management
Platelet transfusion is the mainstay for bleeding episodes. Key considerations:
- Patients develop antibodies to GPIb-IX-V (the antigen they lack) after repeated transfusions, causing refractoriness
- Transfused platelets should be HLA-matched, irradiated, and leukodepleted to minimize alloimmunization risk
- Routine prophylactic transfusion before delivery is controversial due to alloimmune thrombocytopenia (AIT) risk
Additional treatment options:
- Tranexamic acid (antifibrinolytic) - oral, IV, or mouthwash; useful for mucosal bleeds
- DDAVP (desmopressin) - may reduce bleeding in some patients
- Recombinant activated Factor VIIa (rFVIIa) - used in refractory cases or when platelet transfusion fails, especially for surgery or severe bleeds
- Avoid all antiplatelet agents (aspirin, NSAIDs, clopidogrel, etc.)
In pregnancy:
-
Cesarean delivery reserved for obstetric indications only
-
Neuraxial anesthesia should be avoided as normal hemostasis cannot be guaranteed even after prophylactic platelet transfusion
-
Prenatal diagnosis is possible and should be offered to affected families
-
Scott-Brown's Otorhinolaryngology, p. 3605-3616
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 3021-3023
Key Summary Points
- Cause: GPIb-IX-V deficiency → impaired platelet adhesion to vWF
- Genes: GPIBA, GPIBB, GP9 - autosomal recessive
- Triad: Thrombocytopenia + giant platelets + mucocutaneous bleeding
- Lab hallmark: Fails ristocetin only; all other agonists normal (opposite of Glanzmann)
- Confirm with: Flow cytometry (absent GPIbα) ± genetic testing
- Treat with: Platelet transfusions (HLA-matched), tranexamic acid, rFVIIa
Recent literature (2025): A comprehensive review by Kaya Z (2025) covers the full spectrum of BSS from epidemiology and molecular pathology through therapeutic management (PMID: 39191409). A 2026 case report (Okamoto et al.) documents use of rFVIIa as hemostatic rescue in BSS refractory to HLA-matched platelet transfusion (PMID: 41746364), supporting its role in refractory disease.
This is a shared conversation. Sign in to Orris to start your own chat.