Congential myansthenia gravis
Reading File
Finding Sources
Searching PubMed
"congenital myasthenic syndrome"[MeSH Terms]
Finding Sources
Searching PubMed
congenital myasthenic syndrome treatment
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile a thorough response on congenital myasthenic syndromes (CMS).
Congenital Myasthenic Syndromes (CMS)
Note: The user likely means Congenital Myasthenic Syndromes (CMS), not autoimmune myasthenia gravis. These are distinct entities. CMS is a genetic, non-autoimmune disorder of the neuromuscular junction (NMJ).
Definition
Congenital myasthenic syndromes are a heterogeneous group of inherited disorders caused by mutations in genes encoding proteins at the NMJ. Unlike autoimmune myasthenia gravis, CMS is not antibody-mediated - patients do not have antibodies to the nicotinic ACh receptor or MuSK. - Eric Kandel, Principles of Neural Science, 6th Ed., p. 1483
Distinguishing CMS from Other Forms of Childhood Myasthenia
| Type | Etiology | Onset | Sex | Thymus | Course |
|---|---|---|---|---|---|
| Neonatal myasthenia | Passive transfer of maternal antibodies across placenta | Neonatal | Both | Normal | Transient |
| Congenital myasthenia (CMS) | Genetic endplate pathology; autosomal recessive | 0-2 yrs | Male > Female | Normal | Non-fluctuating, compatible with long survival |
| Juvenile myasthenia | Autoimmune | 2-20 yrs | Female > Male (4:1) | Hyperplasia | Relapses/remissions |
- Barash, Cullen & Stoelting's Clinical Anesthesia, 9e, p. 1839
Clinical Features (Common to All CMS Types)
-
Positive family history (autosomal recessive in most forms)
-
Weakness with easy fatigability present since infancy
-
Ptosis (drooping of eyelids)
-
Decremental response to repetitive nerve stimulation on EMG
-
Negative anti-nicotinic ACh receptor antibodies and anti-MuSK antibodies (required for diagnosis)
-
Delayed motor milestones
-
Subnormal skeletal muscle development/bulk
-
Fluctuating ocular palsies brought out by sustained activity
-
Eric Kandel, Principles of Neural Science, p. 1483; Adams & Victor's Principles of Neurology, 12th Ed.
Classification by Site of Defect
CMS is classified into three broad groups based on the location of the molecular defect:
1. Presynaptic Forms
| Syndrome | Gene Defect | Key Features | Treatment |
|---|---|---|---|
| Episodic apnea | Choline acetyltransferase (ChAT) deficiency | Mild weakness; recurrent apnea - life-threatening | AChE inhibitors; apnea monitor |
| Paucity of synaptic vesicles | Unknown | Ptosis; pronounced episodic weakness | AChE inhibitors |
| Reduced quantal ACh release | Unknown | Wasting, respiratory failure, dysmorphism | AChE inhibitors + 3,4-diaminopyridine (3,4-DAP) |
The ChAT deficiency form is notable: the enzyme normally synthesizes ACh from choline + acetyl-CoA, so its absence impairs transmitter synthesis.
2. Synaptic Cleft Forms
| Syndrome | Gene Defect | Key Features | Treatment |
|---|---|---|---|
| AChE deficiency | AChE gene / collagen tail for AChE | Diffuse weakness; prolonged (not reduced) end-plate potentials; repetitive muscle response on EMG | AVOID AChE inhibitors (dangerous) |
| DOK-7 synaptopathy | DOK7 mutation | Limb-girdle weakness; ptosis | None established |
In AChE deficiency, ACh cannot be broken down - end-plate potentials are prolonged, leading to end-plate myopathy from excessive stimulation. Using AChE inhibitors worsens this dramatically. - Eric Kandel, p. 1483
3. Postsynaptic Forms (Most Common)
These arise from mutations in subunits of the nicotinic ACh receptor (AChR) itself:
| Syndrome | Defect | Key Features | Treatment |
|---|---|---|---|
| Slow channel syndrome | AChR subunit mutations (gain-of-function) | Prominent limb weakness (unusual - cranial muscles relatively spared); prolonged channel opening; end-plate myopathy | Quinidine (blocks open channel); AVOID AChE inhibitors |
| Fast channel syndrome | AChR subunit mutations (loss-of-function) | Ptosis; recurrent weakness; dorsal forearm atrophy; motor delay | 3,4-DAP (increases quantal ACh release); AChE inhibitors |
| Primary AChR deficiency | AChR subunit mutations (reduced expression) | Ptosis; recurrent weakness | AChE inhibitors + 3,4-DAP |
| Rapsyn deficiency | RAPSN mutation | Ptosis; recurrent weakness (rapsyn clusters AChR) | AChE inhibitors + 3,4-DAP |
| Plectin deficiency | Plectin | Myasthenic features + epidermolysis bullosa | 3,4-DAP |
| Escobar syndrome | Fetal gamma-AChR subunit | Arthrogryposis, respiratory failure, pterygium (skin webbing) - in utero onset | Improves with maturity as fetal gamma-subunit replaced by epsilon-subunit |
- Adams & Victor's Principles of Neurology, 12th Ed.
Pathophysiology Key Points
- Slow channel syndrome: Mutations increase AChR affinity for ACh and slow channel closing. Prolonged opening leads to cation overload and end-plate myopathy. Same danger as AChE deficiency - AChE inhibitors are potentially harmful.
- Fast channel syndrome: Mutations accelerate channel closing, reducing depolarization time. 3,4-DAP helps by blocking presynaptic K+ channels, prolonging the action potential, and increasing quantal ACh release.
- Neonatal myasthenia (passive) resolves spontaneously within weeks as maternal antibodies are cleared. CMS persists lifelong.
Diagnosis
- Clinical suspicion - fatigable weakness since birth/infancy, family history
- Serological testing - absence of anti-AChR, anti-MuSK, anti-LRP4 antibodies (required)
- EMG - decremental response to repetitive stimulation; repetitive CMAP in AChE deficiency
- Edrophonium (Tensilon) test - inconsistently positive; positive in some forms (fast channel, rapsyn deficiency), negative or harmful in others (AChE deficiency, slow channel)
- Genetic testing - definitive; whole exome sequencing is the modern gold standard
- Muscle biopsy / electron microscopy - can show end-plate morphology, synaptic vesicle paucity
- Bradley & Daroff's Neurology in Clinical Practice; Adams & Victor's
Treatment Summary
| Drug | Mechanism | Use in CMS |
|---|---|---|
| AChE inhibitors (pyridostigmine) | Prevent ACh breakdown | Useful in presynaptic forms, fast channel, rapsyn/AChR deficiency; AVOID in AChE deficiency and slow channel syndrome |
| 3,4-Diaminopyridine (3,4-DAP) | Blocks presynaptic K+ channels → prolongs AP → more ACh release | Useful in fast channel, rapsyn, plectin deficiencies |
| Quinidine | Blocks open slow channels | Specific for slow channel syndrome |
Unlike autoimmune MG, CMS does not respond to immunosuppressants, plasmapheresis, or thymectomy.
Recent Literature
Recent reviews confirm the genetic heterogeneity of CMS with over 30 causative genes now identified. Key recent publications:
- Beeson D (2024). Congenital myasthenic syndromes. Handbook of Clinical Neurology - comprehensive current review [PMID: 39174255]
- Spendiff S et al (2025). Congenital myasthenic syndromes. Int Rev Neurobiol - covers emerging genetic subtypes [PMID: 40675739]
- Wiendl H et al (2023). Guidelines for the management of myasthenic syndromes. Therapeutic Advances in Neurological Disorders - includes CMS management [PMID: 38152089]
Key Takeaways
- CMS = genetic, not autoimmune; antibody-negative
- Must distinguish from neonatal (transient, maternal antibodies) and juvenile (autoimmune) forms
- Three anatomical subtypes: pre- / synaptic / postsynaptic - each with distinct mutations and treatment implications
- Critical pearl: AChE inhibitors are contraindicated in AChE deficiency and slow channel syndrome (worsen end-plate myopathy)
- Genetic confirmation is now feasible with next-generation sequencing and is the standard of care
What are the monoclonal antibodies are used in the t cell all
Reading File
Finding Sources
Finding Sources
Searching PubMed
"T-cell ALL" OR "T-cell acute lymphoblastic leukemia" monoclonal antibody
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Searching PubMed
"T-ALL" immunotherapy monoclonal antibody CD7 CD38
Reading Article
PMID: 41348009
Reading Article
PMID: 42165843
I now have comprehensive, current information. Here is the full answer:
Monoclonal Antibodies in T-Cell ALL (T-ALL)
Important Context First
T-ALL presents a unique challenge for immunotherapy. Unlike B-ALL, where a rich arsenal of monoclonal antibodies (rituximab, blinatumomab, inotuzumab) is well-established, T-ALL lacks approved targeted antibody therapies as of the current standard of care. This is because many T-cell antigens (CD3, CD7, CD5) are shared between the malignant blasts AND the normal T cells or the CAR-T cells used for therapy - creating fratricide and immunosuppression problems. - Harrison's Principles of Internal Medicine 22E, p. 892
As Harrison's states directly: "Immunotherapy for T-ALL is still not available, and intensive chemotherapy is still the mainstay in combination with the T cell-specific drug nelarabine." - Harrison's 22E
Agents Currently Used or Under Investigation in T-ALL
1. Blinatumomab (Anti-CD19 x Anti-CD3 BiTE)
- Type: Bispecific T-cell engager (BiTE) - not a classic monoclonal antibody
- Mechanism: Links CD3 on T cells to CD19 on leukemic blasts - recruits the patient's own T cells to kill CD19+ cells
- T-ALL use: CD19 is variably expressed in T-ALL (unlike B-ALL where it is nearly universal). Its use in T-ALL is therefore limited and investigational
- Primarily established in B-ALL (MRD+ and relapsed/refractory settings)
- Goldman-Cecil Medicine, p. 1925
2. Nelarabine (not a monoclonal antibody - important distinction)
- A nucleoside analogue prodrug converted to ara-GTP, toxic to T cells
- The only T-ALL-specific agent approved by FDA (for relapsed/refractory T-ALL)
- Achieves ~30% CR rate as a single agent in recurrent T-ALL
- Often combined with standard regimens (e.g., COG AALL0434 pediatric protocol)
- Goldman-Cecil Medicine, p. 1925
Investigational Monoclonal Antibodies Targeting T-ALL Antigens
These are under active clinical/preclinical investigation as of 2025-2026:
| Target | Agent | Notes |
|---|---|---|
| CD38 | Daratumumab (anti-CD38) | CD38 is expressed on T-ALL blasts; daratumumab being explored in frontline combinations; promising early data in R/R T-ALL. Boissel N, ASH 2025 |
| CD7 | Anti-CD7 CAR-T cells / anti-CD7 antibody-drug conjugates | CD7 expressed on ~95% T-ALL blasts but also on normal T cells - fratricide challenge; CAR-T cells being engineered to knock out CD7 first |
| CD5 | Anti-CD5 CAR-T cells | CD5 expressed on most T-ALL; CAR-T cell approaches under study |
| CD52 | Alemtuzumab (anti-CD52) | CD52 expressed on all B and T lymphocytes including T-ALL; used in some protocols (e.g., UKALL2003); mainly in pediatric ALL as CNS-directed or MRD-directed therapy |
| CD2 | Anti-CD2 agents | Investigational |
| PD-1 | Nivolumab, Pembrolizumab | Checkpoint inhibitors; being explored in combination with chemotherapy in R/R T-ALL |
| CTLA-4 | Ipilimumab | Explored in combination immunotherapy regimens |
Sources: Harrison's 22E; Boissel 2025 [PMID 41348009]; Celona et al. 2026 [PMID 42165843]
Surface Antigens of T-ALL (Diagnostic Markers, Not All Therapeutic Targets)
T-ALL blast cells express the following markers, which define the immunological subtype:
| Marker | Expression in T-ALL | Clinical Significance |
|---|---|---|
| CD7 | ~95-100% | Most consistent T-ALL marker; therapeutic target under development |
| CyCD3 / sCD3 | All cases | Definitive T-lineage marker |
| CD2 | Variable | E-rosette receptor |
| CD1a | Cortical/thymic T-ALL | Absent in ETP-ALL |
| CD5 | Most (weak in ETP-ALL) | Distinguishes ETP-ALL |
| CD8, CD4 | Variable | Stage of differentiation |
| CD10 | Some | Also in B-ALL |
| CD34 | ETP-ALL | Stem cell marker |
Harrison's 22E, p. 892
Why T-ALL Immunotherapy Lags Behind B-ALL
| Issue | Detail |
|---|---|
| Antigen sharing | CD3, CD7, CD5 are expressed on both malignant T cells AND the normal T cells used in CAR-T manufacturing |
| Fratricide | Anti-T cell CAR-T constructs kill each other during manufacturing |
| Immunosuppression | Targeting T-cell antigens depletes the immune system |
| CD19 absence | Blinatumomab (most effective BiTE in ALL) targets CD19, which is not reliably expressed in T-ALL |
Summary
| Category | Agent | Status in T-ALL |
|---|---|---|
| Approved, T-ALL specific | Nelarabine (not a mAb) | FDA-approved for R/R T-ALL |
| Investigational mAb | Daratumumab (anti-CD38) | Phase I/II trials; promising |
| Investigational cellular | Anti-CD7 CAR-T, Anti-CD5 CAR-T | Early trials |
| BiTE (limited role) | Blinatumomab | Only if CD19+ T-ALL |
| Non-specific lymphocyte | Alemtuzumab (anti-CD52) | Some protocols; not T-ALL specific |
| Checkpoint inhibitors | Nivolumab, Pembrolizumab | Investigational combinations |
The most active area of current research is anti-CD38 (daratumumab) and CAR-T cells engineered against CD7 or CD5 with fratricide-prevention strategies. As of 2025-2026, none of these have regulatory approval specifically for T-ALL. - Boissel N, 2025 | Celona et al., 2026
Can I use the rituximab for the t cell all
Finding Sources
Finding Sources
Loading Image
Reading File
The textbooks confirm this clearly. Here is the definitive answer:
Can Rituximab Be Used in T-Cell ALL?
The Short Answer: No
Rituximab cannot be used in T-cell ALL. Here is exactly why:
Why Rituximab Does Not Work in T-ALL
Rituximab is a chimeric anti-CD20 monoclonal antibody. It works by binding to the CD20 antigen on cell surfaces and triggering cell death via:
- Complement-dependent cytotoxicity (CDC)
- Antibody-dependent cellular cytotoxicity (ADCC)
- Direct apoptosis induction
The fundamental problem: CD20 is a B-cell antigen. It is expressed on B-lineage cells across all stages of B-cell development, but it is absent on T cells at every stage of differentiation.
T-Cell Surface Markers (What T-ALL Actually Expresses)
Looking at the T-cell differentiation diagram from Harrison's 22E:
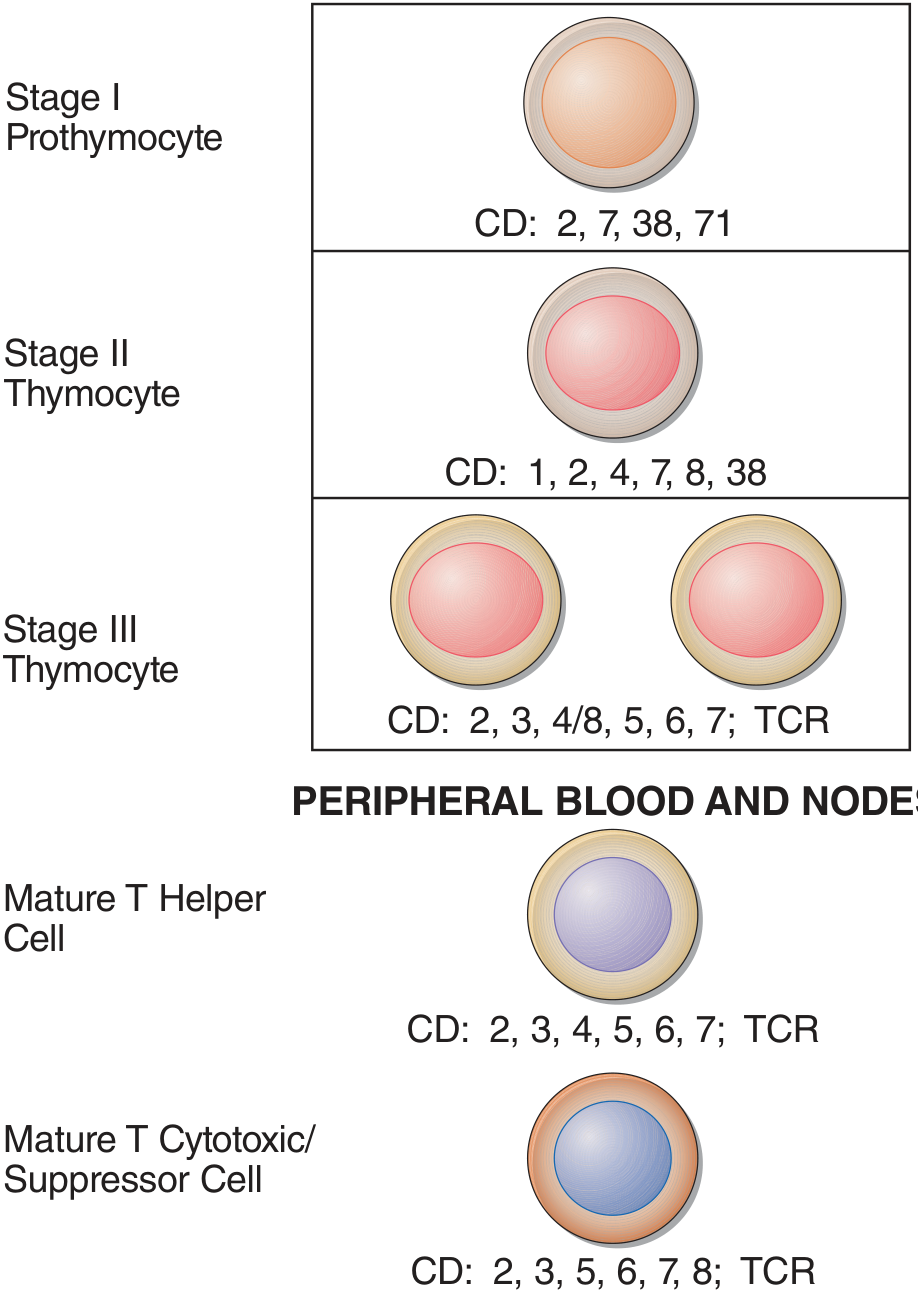
T-ALL blasts express CD2, CD3, CD4, CD5, CD7, CD8, CD38 - at various stages. CD20 is conspicuously absent from all stages of T-cell development. - Harrison's Principles of Internal Medicine 22E, p. 903
Dermatology textbook confirms the immunophenotype contrast directly:
- B-cell ALL: CD19+, TdT+, CD20+, CD5+, CD10+, CD22+, CD79a+, PAX-5+
- T-cell ALL: TdT+, CD1a+, CD2+, CD3c+, CD4+, CD5+, CD7+, CD8+ ← No CD20
Where Rituximab IS Used in ALL
Rituximab is used only in B-lineage ALL, specifically:
| ALL Subtype | CD20 Expression | Rituximab Use |
|---|---|---|
| Burkitt's leukemia/lymphoma (mature B-ALL) | 86-100% | YES - dramatically improves OS to >80% |
| B-precursor ALL | 30-40% | YES - included in most B-ALL regimens (375 mg/m², day -1, ≥8 cycles) |
| T-cell ALL | 0% - not expressed | NO |
Harrison's 22E, p. 892 - "Anti-CD20: The anti-CD20 monoclonal antibody rituximab has improved the outcome of patients with de novo Burkitt's leukemia/lymphoma... Rituximab is now included in most B-ALL regimens."
What IS Used in T-ALL Instead
Since rituximab has no role, T-ALL treatment relies on:
| Drug | Type | Role |
|---|---|---|
| Nelarabine | Nucleoside analogue (NOT a mAb) | Only T-ALL-specific approved agent; ~30% CR in relapsed T-ALL |
| Daratumumab (anti-CD38) | Monoclonal antibody | Investigational; CD38 IS expressed on T-ALL (see image above - Stage I and II thymocytes) |
| Alemtuzumab (anti-CD52) | Monoclonal antibody | Some protocols; targets CD52 on all lymphocytes |
| Standard chemotherapy | Multi-agent | Still the mainstay of T-ALL treatment |
Memory Hook
Rituximab = anti-CD20 = B-cells only T-ALL expresses CD2, 3, 5, 7, 38 - never CD20 Therefore rituximab has no target in T-ALL and is not used.
This is a shared conversation. Sign in to Orris to start your own chat.