Explain the mechanism of acute respiratory distress syndrome
Reading File
search_medical_text
ARDS acute respiratory distress syndrome pathophysiology mechanism
search_medical_text
ARDS diffuse alveolar damage phases exudative proliferative fibrotic
GradeResponse
GradeResponse
search_medical_text
ARDS exudative phase neutrophil cytokine alveolar epithelial injury pulmonary edema
search_medical_image
ARDS pathophysiology diffuse alveolar damage mechanism diagram
GradeResponse
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Core Pathophysiologic Features
ARDS is a syndrome defined by three cardinal pathophysiologic features (ARDS Definition Guidelines, p. 2):
- Increased alveolo-capillary membrane permeability → inflammatory (non-cardiogenic) pulmonary edema
- Increased non-aerated lung tissue → reduced lung compliance (higher elastance)
- Increased venous admixture and dead space → hypoxemia (low PaO₂/FiO₂ ratio) and hypercapnia
Initiating Triggers
ARDS arises from either direct (pulmonary) or indirect (extrapulmonary) insults:
| Direct | Indirect |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis |
| Aspiration of gastric contents | Severe trauma / shock |
| Pulmonary contusion | Pancreatitis |
| Inhalation injury | Massive transfusion (TRALI) |
| Near-drowning | Burns |
The Three Phases of ARDS
Harrison's Principles of Internal Medicine (p. 8196) describes a natural history of three overlapping phases, each with distinct cellular and structural features:
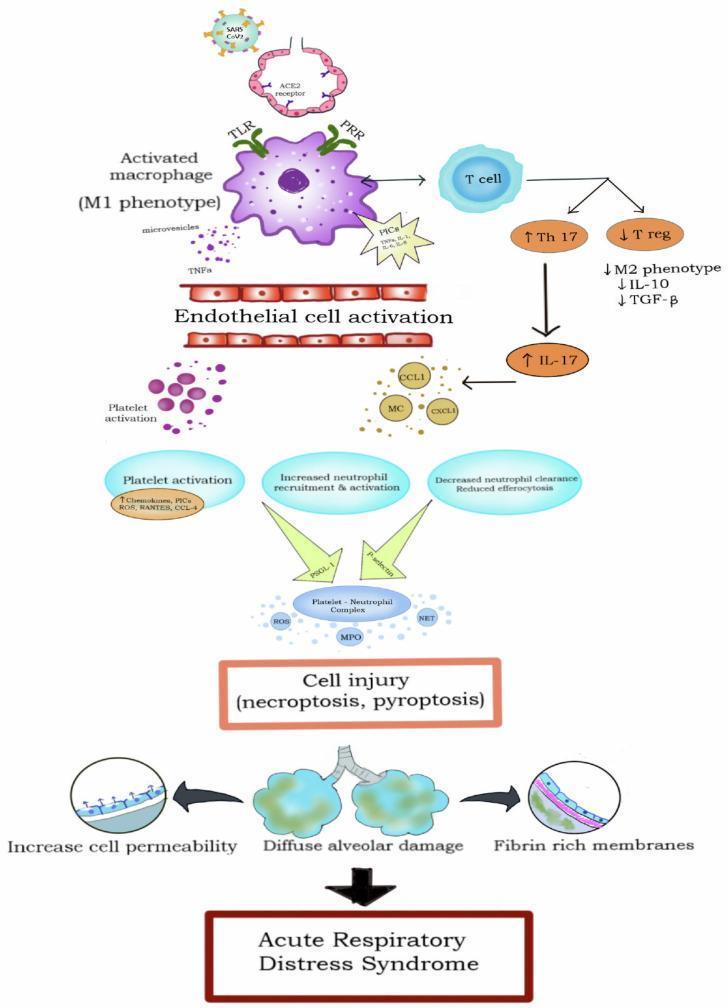
Phase 1 — Exudative Phase (Days 1–7)
This is the dominant inflammatory phase:
- Initial insult activates alveolar macrophages, which release pro-inflammatory cytokines: TNF-α, IL-1β, IL-6, IL-8.
- These cytokines recruit neutrophils from the pulmonary capillaries into the alveolar space.
- Activated neutrophils release:
- Reactive oxygen species (ROS)
- Proteases (elastase, matrix metalloproteinases)
- Myeloperoxidase (MPO)
- Neutrophil extracellular traps (NETs)
- This toxic arsenal directly injures the alveolar epithelium (type I pneumocytes) and vascular endothelium.
Consequences of epithelial and endothelial injury:
- Loss of tight junctions → protein-rich fluid floods the alveolar space (non-cardiogenic pulmonary edema)
- Destruction of type II pneumocytes → impaired surfactant production → alveolar collapse (atelectasis)
- Formation of hyaline membranes (fibrin + cellular debris lining the denuded alveoli) — the histologic hallmark of diffuse alveolar damage (DAD)
- Platelet-neutrophil complexes (via P-selectin/PSGL-1 interactions) amplify microvascular injury
Physiologic result: Flooded, collapsed alveoli create intrapulmonary shunt → refractory hypoxemia. The non-aerated but perfused lung units are unresponsive to supplemental oxygen alone.
Phase 2 — Proliferative Phase (Days 7–21)
If the patient survives:
- Type II pneumocytes proliferate to resurface the denuded alveolar epithelium and differentiate into type I cells.
- Inflammation begins to resolve; macrophage phenotype shifts toward anti-inflammatory (M2).
- Fibroblast migration and proliferation begins — early fibroproliferation.
- Edema fluid is reabsorbed as the epithelial barrier is restored.
Most patients improve during this phase, though lung mechanics remain abnormal.
Phase 3 — Fibrotic Phase (Weeks to Months)
A subset of patients progress to:
- Extensive fibrosis of the alveolar walls, interstitium, and airspaces
- Architectural distortion of lung parenchyma
- Obliteration of the pulmonary microvasculature → pulmonary hypertension
- Cystic changes and a "honeycomb" pattern on imaging
- Persistent dead space ventilation → hypercapnia and ventilator dependence
The stimulus for fibrosis appears to be sustained TGF-β signaling and disordered repair. This phase carries the highest mortality and longest ICU course.
Key Molecular Mediators Summary
| Mediator | Source | Effect |
|---|---|---|
| TNF-α, IL-1β, IL-6 | Macrophages | Neutrophil recruitment, systemic inflammation |
| IL-8 (CXCL8) | Epithelium, macrophages | Potent neutrophil chemoattractant |
| ROS, MPO | Neutrophils | Direct oxidative tissue injury |
| NETs | Neutrophils | Endothelial/epithelial damage, thrombosis |
| TGF-β | Multiple | Fibroproliferation, impaired epithelial repair |
| Surfactant deficit | Injured type II cells | Alveolar collapse, reduced compliance |
Physiologic Consequences
- Reduced PaO₂/FiO₂ ratio (Berlin criteria: <300 mmHg with PEEP ≥5 cmH₂O)
- Reduced lung compliance — "stiff lungs" requiring high airway pressures
- Intrapulmonary shunting — perfusion of non-ventilated alveoli
- Increased dead space — ventilation of non-perfused units → CO₂ retention
- Pulmonary hypertension — hypoxic vasoconstriction + vascular remodeling → right ventricular strain
Summary
ARDS is fundamentally a syndrome of uncontrolled innate immune activation causing diffuse alveolar damage. The inciting insult — whether pulmonary or systemic — activates macrophages and recruits neutrophils, whose cytotoxic arsenal destroys the alveolo-capillary barrier. The result is flooding of alveoli with protein-rich edema, loss of surfactant, and refractory hypoxemia. Recovery depends on the balance between further injury and epithelial repair; fibroproliferation in a subset leads to irreversible lung remodeling.
This is a shared conversation. Sign in to Orris to start your own chat.