I now have extensive, high-quality content from authoritative medical textbooks to answer all 17 questions comprehensively. Let me compile the full detailed answers.
Answers to All 17 Questions on Hematological Malignancies
1. Commonest Childhood Malignancy
Acute Lymphoblastic Leukemia (ALL) - specifically B-cell ALL (B-ALL) - is the commonest childhood malignancy. It has a peak incidence between ages 2 and 10 years. ALL is the most frequent neoplastic disease in children, with an early peak at age 3-4 years. More than 80% of children with B-ALL and T-ALL are cured, making it one of oncology's great success stories.
(Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22E)
2. Two Genes Identified as Risk Factors for ALL
The two most important genes/mutations identified as risk factors for ALL are:
-
BCR-ABL fusion gene (Philadelphia chromosome - t(9;22)): Present in ~5% of childhood B-ALL and ~25% of adult B-ALL; associated with worse prognosis. In the elderly, Ph+ ALL accounts for half of B-lineage cases.
-
KMT2A (MLL) gene rearrangements: Associated with infantile ALL (< 2 years of age) and carries a particularly poor prognosis. Translocations of KMT2A produce aggressive leukemias that are difficult to treat.
Additional risk factor genes include PAX5 (required for B-cell differentiation; commonly mutated in B-ALL) and IKZF1 (Ikaros), but BCR-ABL and KMT2A are the two most classically cited.
(Robbins & Kumar Basic Pathology; Harrison's 22E)
3. One Classification System of ALL
The World Health Organization (WHO) Classification is the widely accepted system for ALL. It classifies ALL based on:
- Immunophenotype: B-lymphoblastic leukemia/lymphoma vs. T-lymphoblastic leukemia/lymphoma
- Cytogenetics/molecular genetics: e.g., B-ALL with t(9;22)/BCR-ABL, B-ALL with t(v;11q23)/KMT2A, B-ALL with t(12;21)/ETV6-RUNX1 (TEL-AML1), B-ALL with hyperdiploidy, hypodiploidy, etc.
(The older FAB [French-American-British] classification used morphology alone [L1, L2, L3] but has largely been replaced by the WHO classification)
(Robbins & Kumar Basic Pathology)
4. Most Definitive Method of Distinguishing B-Cell and T-Cell Lineages
Immunophenotyping by flow cytometry (or immunohistochemistry) is the most definitive method. It uses antibodies against lineage-specific surface markers:
- B-cell markers: CD19, CD20, CD22, CD10, surface/cytoplasmic immunoglobulin
- T-cell markers: CD2, CD3, CD5, CD7, CD4, CD8
- Both ALL types express: TdT (terminal deoxynucleotidyl transferase) - a specialized DNA polymerase expressed in pre-B and pre-T cells
"Definitive diagnosis relies on stains performed with antibodies specific for B, T, and myeloid lineage-specific markers, usually by flow cytometry." - Robbins & Kumar Basic Pathology
(Robbins & Kumar Basic Pathology)
5. Favorable-Risk Genetics for AML
Favorable-risk cytogenetics/molecular genetics in AML include:
| Genetic Finding | Mechanism |
|---|
| t(15;17) - PML-RARA | Acute promyelocytic leukemia (APL); responds to ATRA + arsenic trioxide; very high cure rates |
| t(8;21) - RUNX1-RUNX1T1 | Favorable prognosis; responds well to high-dose cytarabine |
| inv(16) or t(16;16) - CBFB-MYH11 | Core-binding factor AML; favorable prognosis |
| NPM1 mutation (without FLT3-ITD) | Favorable prognosis |
| Biallelic CEBPA mutation | Favorable prognosis |
The core-binding factor AMLs [t(8;21) and inv(16)] and APL are the classic favorable-risk groups.
(Robbins & Kumar Basic Pathology; Harrison's 22E)
6. High-Risk Genetics for AML
High-risk (adverse) genetics for AML include:
| Genetic Finding | Comment |
|---|
| Complex karyotype (≥3 abnormalities) | Very poor prognosis |
| Monosomal karyotype | Worst prognosis |
| TP53 mutations | Dismal outcome even with stem cell transplantation |
| FLT3-ITD (especially high allelic ratio) | Associated with high relapse rate |
| ASXL1 mutations | Poor prognosis, especially in older patients |
| inv(3)/t(3;3) - GATA2-MECOM | Poor prognosis |
| del(5q), del(7q), -5, -7 | Poor prognosis |
| KMT2A (MLL) rearrangements | Poor prognosis |
| Secondary AML (post-MDS/MPN or therapy-related) | Poor prognosis |
(Harrison's 22E; Robbins & Kumar Basic Pathology)
7. Complications of Hematological Malignancy
Key complications include:
From marrow failure:
- Anemia (fatigue, weakness)
- Thrombocytopenia (petechiae, bleeding)
- Neutropenia (recurrent/opportunistic infections)
Disease-specific:
- DIC (Disseminated Intravascular Coagulation): Especially in AML-M3 (APL), where promyelocytes release thromboplastic substances
- Tumor lysis syndrome: Hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia - can cause acute renal failure
- Leukostasis: In high blast counts (>100,000/μL) - can cause pulmonary/CNS symptoms
- Hyperleukocytosis
- CNS involvement (meningeal leukemia - especially ALL)
- Mediastinal compression: T-ALL with thymic mass
- Hemophagocytic lymphohistiocytosis (HLH): Fever, splenomegaly, pancytopenia, very high ferritin (>10,000 μg/L)
- Hyperviscosity syndrome (multiple myeloma, Waldenström's)
- Spinal cord compression (lymphoma)
- Treatment-related: Secondary malignancies, cardiotoxicity from anthracyclines, therapy-related AML
(Robbins & Kumar Basic Pathology)
8. Four Stages of ALL Chemotherapy
ALL chemotherapy consists of 4 sequential phases:
-
Induction - Goal: achieve complete remission (eliminate >99% of leukemic cells). Uses vincristine, corticosteroids (prednisone/dexamethasone), L-asparaginase ± anthracycline.
-
Consolidation (Intensification) - Goal: eliminate residual disease. Uses high-dose methotrexate, high-dose cytarabine, or re-induction-like cycles.
-
CNS Prophylaxis - Intrathecal chemotherapy (methotrexate ± cytarabine) ± cranial irradiation. Essential because the CNS is a "sanctuary site" not penetrated by systemic drugs.
-
Maintenance - Goal: prevent relapse. Uses oral 6-mercaptopurine (6-MP) + methotrexate weekly for 2-3 years.
(Goldman-Cecil Medicine; Harrison's 22E)
9. Philadelphia Chromosome-Positive ALL
- Definition: t(9;22)(q34;q11) translocation creating the BCR-ABL fusion gene - the "Philadelphia chromosome"
- Frequency: ~5% of childhood ALL; 25% of adult B-ALL; >50% in elderly B-lineage ALL
- Mechanism: BCR-ABL encodes a constitutively active tyrosine kinase that activates all downstream growth factor receptor pathways, driving proliferation and survival
- Prognosis: Historically very poor; now dramatically improved with tyrosine kinase inhibitors (TKIs) such as imatinib, dasatinib, or ponatinib combined with chemotherapy
- Treatment: TKI (dasatinib/ponatinib) + chemotherapy, followed by allogeneic hematopoietic stem cell transplantation in eligible patients
(Robbins & Kumar Basic Pathology; Harrison's 22E)
10. What % of Transient Myeloproliferative Disorder (TMD) Develops AML?
- ~20-30% of infants with Down syndrome (trisomy 21) who have Transient Myeloproliferative Disorder (TMD, also called Transient Abnormal Myelopoiesis/TAM) will progress to acute megakaryoblastic leukemia (AML-M7) within the first 4 years of life.
- TMD itself resolves spontaneously in most cases within 3 months, but the ~20-30% subset with GATA1 mutations progresses to AML.
- Harrison's notes Down syndrome patients have a 20-fold increased incidence of leukemia overall.
(Harrison's 22E)
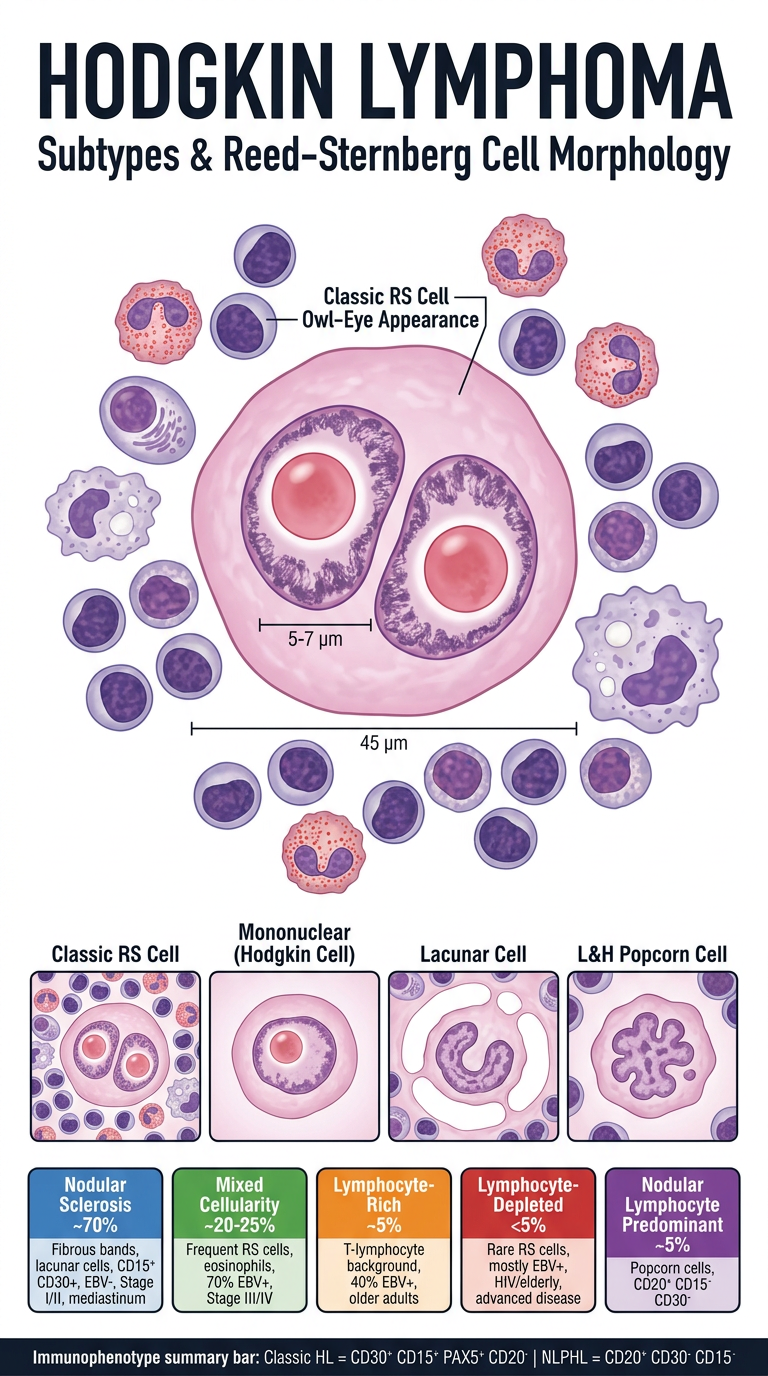
11. WHO Classification of Hodgkin Lymphoma
The WHO classifies Hodgkin Lymphoma into 5 subtypes:
- Nodular Sclerosis (most common - ~70%)
- Mixed Cellularity (~20-25%)
- Lymphocyte-Rich (~5%)
- Lymphocyte-Depleted (rare, <5%)
- Nodular Lymphocyte Predominant (~5%)
Subtypes 1-4 are grouped as Classic Hodgkin Lymphoma (cHL) - Reed-Sternberg cells are CD15+, CD30+, PAX5+, CD20-.
Subtype 5 (Nodular Lymphocyte Predominant) has a different immunophenotype: CD20+, CD15-, CD30-, and is now sometimes renamed "nodular lymphocyte-predominant B-cell lymphoma" in the International Consensus Classification.
(Robbins, Cotran & Kumar Pathologic Basis of Disease)
12. Hallmark of Classic Hodgkin Lymphoma
The hallmark is the Reed-Sternberg (RS) cell - a neoplastic giant cell present against a background of reactive inflammatory cells (lymphocytes, eosinophils, plasma cells, macrophages which make up >90% of the tumor cellularity).
Immunophenotype of classic RS cells:
- CD30+ (90-100% of cases)
- CD15+ (75-85% of cases)
- PAX5+ (dim, B-cell transcription factor)
- CD20- (negative)
- EBV present in a subset
The RS cells derive from germinal center or post-germinal center B cells but paradoxically fail to express most B-cell genes including immunoglobulin genes - this silencing is caused by widespread epigenetic changes.
(Robbins, Cotran & Kumar Pathologic Basis of Disease; Goldman-Cecil Medicine)
13. Description of a Reed-Sternberg Cell
Reed-Sternberg cells are large binucleated or multinucleated giant cells (~45 μm in diameter) with:
- Two or more nuclear lobes (or a bilobed nucleus) - giving an "owl-eye" appearance
- Large inclusion-like nucleoli (~5-7 μm - about the size of a small lymphocyte) - one prominent nucleolus per lobe
- Abundant cytoplasm
Variants recognized:
| Variant | Description | Subtype |
|---|
| Mononuclear (Hodgkin cell) | Single nucleus with large inclusion-like nucleolus | Classic HL |
| Lacunar cell | Folded/multilobate nucleus, pale cytoplasm; sits in an empty space (lacuna = artifact of formalin fixation) | Nodular sclerosis |
| "Mummified" cell | Pyknotic, shrunken - undergone cell death | Classic HL |
| L&H "popcorn" cell (lymphohistiocytic) | Polypoid nucleus, inconspicuous nucleoli, moderate pale cytoplasm | Nodular lymphocyte predominant |
(Robbins, Cotran & Kumar Pathologic Basis of Disease)
14. Four Varieties of Classic Hodgkin Lymphoma
| Subtype | Key Features | Clinical |
|---|
| 1. Nodular Sclerosis | Lacunar RS cells; fibrous bands divide tissue into nodules; CD15+, CD30+; usually EBV- | Most common (~70%); usually Stage I/II; mediastinal involvement; equal M:F; young adults |
| 2. Mixed Cellularity | Frequent mononuclear and diagnostic RS cells; rich in lymphocytes, eosinophils, plasma cells; CD15+, CD30+; ~70% EBV+ | >50% present as Stage III/IV; male > female; bimodal age distribution |
| 3. Lymphocyte-Rich | Frequent mononuclear and diagnostic RS cells; background rich in T lymphocytes; CD15+, CD30+; ~40% EBV+ | Uncommon; male > female; older adults |
| 4. Lymphocyte-Depleted | Frequent RS cells with few reactive cells; CD15+, CD30+; mostly EBV+ | Uncommon; older males, HIV-infected, low-resource countries; often advanced disease |
(Robbins, Cotran & Kumar Pathologic Basis of Disease - Table 13.8)
15. Staging for Hodgkin Lymphoma
Ann Arbor Staging Classification (used for both HL and NHL):
| Stage | Definition |
|---|
| Stage I | Involvement of a single lymph node region (I) or single extralymphatic organ/site (IE) |
| Stage II | Involvement of ≥2 lymph node regions on the SAME side of the diaphragm (II), or with localized extralymphatic involvement (IIE) |
| Stage III | Involvement of lymph node regions on BOTH sides of the diaphragm without (III) or with (IIIE) localized extralymphatic involvement |
| Stage IV | Diffuse/disseminated involvement of ≥1 extralymphatic organ/site (e.g., liver, bone marrow, lung) with or without lymphatic involvement |
Additional suffixes:
- A = absence of systemic symptoms
- B = presence of: unexplained fever, drenching night sweats, and/or unexplained weight loss >10% of body weight ("B symptoms")
- E = extralymphatic involvement
- X = bulky disease
(Robbins, Cotran & Kumar Pathologic Basis of Disease - Table 13.9; Ann Arbor Classification)
16. Four Types of NHL
The four most common types of Non-Hodgkin Lymphoma are:
- Diffuse Large B-Cell Lymphoma (DLBCL) - ~30% of all NHL; most common overall; aggressive
- Follicular Lymphoma - ~20% of all NHL; second most common; indolent; more frequent in North America/Western Europe
- Marginal Zone/MALT Lymphoma - ~5-10%; associated with chronic inflammation (e.g., H. pylori, Sjögren's); indolent
- Mantle Cell Lymphoma - ~5-10%; aggressive; t(11;14) cyclin D1 overexpression
(Other common types include: Small Lymphocytic Lymphoma/CLL, Burkitt Lymphoma, Peripheral T-Cell Lymphoma, Anaplastic Large Cell Lymphoma)
(Goldman-Cecil Medicine)
17. Classification of NHL
The 2016 WHO Classification (updated) is the current standard. It divides NHLs by:
A. Cell of origin:
B-cell neoplasms (85-90% of NHL in US/Europe):
- Precursor: B-lymphoblastic leukemia/lymphoma
- Mature/peripheral B-cell:
- Diffuse large B-cell lymphoma (DLBCL)
- Follicular lymphoma
- Mantle cell lymphoma (t(11;14))
- Marginal zone lymphoma (nodal, splenic, extranodal/MALT)
- Small lymphocytic lymphoma (SLL)/CLL
- Burkitt lymphoma (t(8;14), c-MYC)
- Primary mediastinal (thymic) large B-cell lymphoma
- Plasma cell neoplasms (myeloma)
T/NK-cell neoplasms (~10-15%):
- Precursor: T-lymphoblastic leukemia/lymphoma
- Mature/peripheral T-cell:
- Peripheral T-cell lymphoma, NOS
- Anaplastic large cell lymphoma (ALCL)
- Angioimmunoblastic T-cell lymphoma
- Adult T-cell leukemia/lymphoma (HTLV-1)
- Mycosis fungoides/Sézary syndrome (cutaneous)
- Hepatosplenic T-cell lymphoma
Key principle: Classification integrates morphology + immunophenotype (CD markers) + cytogenetics/molecular genetics + clinical features. About 85% of NHL can be accurately classified by experienced hematopathologists using WHO criteria with adequate material.
(Goldman-Cecil Medicine; Robbins & Kumar Basic Pathology)
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease | Robbins & Kumar - Basic Pathology | Harrison's Principles of Internal Medicine 22E (2025) | Goldman-Cecil Medicine International Edition