teach me full concept of atherosclerosis pathology mbbs
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the textbook content and images needed. Let me compile the comprehensive MBBS-level answer on atherosclerosis pathology.
Atherosclerosis - Complete Pathology for MBBS
Sources: Robbins & Kumar Basic Pathology 10e | Robbins, Cotran & Kumar Pathologic Basis of Disease 10e
1. Definition & Overview
Atherosclerosis (from Greek: athero = gruel/porridge + sclerosis = hardening) is a chronic inflammatory disease of large and medium-sized arteries characterized by intimal lesions called atheromas (atherosclerotic plaques) - raised lesions composed of:
- A soft, friable lipid core (mainly cholesterol, cholesterol esters, necrotic debris)
- Covered by a fibrous cap
It underlies coronary, cerebral, and peripheral vascular disease, and causes roughly half of all deaths in Western countries. Myocardial infarction alone is responsible for ~25% of all deaths in the United States.
2. Risk Factors
Non-Modifiable (Constitutional)
| Factor | Details |
|---|---|
| Genetics | Most important independent risk factor. Familial hypercholesterolemia (LDL receptor mutations) causes MI before age 20 in homozygotes |
| Age | Progressive process; symptomatic disease typically emerges after age 40 in men |
| Sex | Males at higher risk; females gain equal risk post-menopause (estrogen is atheroprotective) |
Modifiable (Major)
| Factor | Mechanism / Impact |
|---|---|
| Hyperlipidemia | Elevated LDL is the dominant lipid risk. Lowering cholesterol slows progression and causes partial plaque regression |
| Hypertension | Increases IHD risk ~60% vs. normotensives; mechanical stress causes endothelial injury |
| Cigarette smoking | Prolonged use doubles IHD death rate; cessation substantially reduces risk |
| Diabetes mellitus | 2x MI risk; 100x increased risk of atherosclerotic gangrene in lower extremities; induces hypercholesterolemia |
Additional Risk Factors
- Inflammation / CRP - CRP is a strong independent marker of risk for MI, stroke, and sudden cardiac death (even in apparently healthy people). CRP levels fall with statins, exercise, and weight loss.
- Hyperhomocysteinemia - Correlates with coronary, peripheral, and cerebrovascular disease
- Metabolic syndrome - Central obesity + insulin resistance + hypertension + dyslipidemia + hypercoagulability
- Lipoprotein(a) [Lp(a)] - Elevated levels independently raise coronary and cerebrovascular risk
- Hemostatic factors - Elevated plasminogen activator inhibitor-1 (PAI-1) predicts MI and stroke
Note: ~20% of cardiovascular events occur in people with none of the classic major risk factors. More than 75% of cardiovascular events in previously healthy females occur with LDL < 160 mg/dL.
3. Pathogenesis - "Response to Injury" Hypothesis
The currently accepted view: atherosclerosis is a chronic inflammatory response of the arterial wall to endothelial injury. Proposed by Russell Ross.
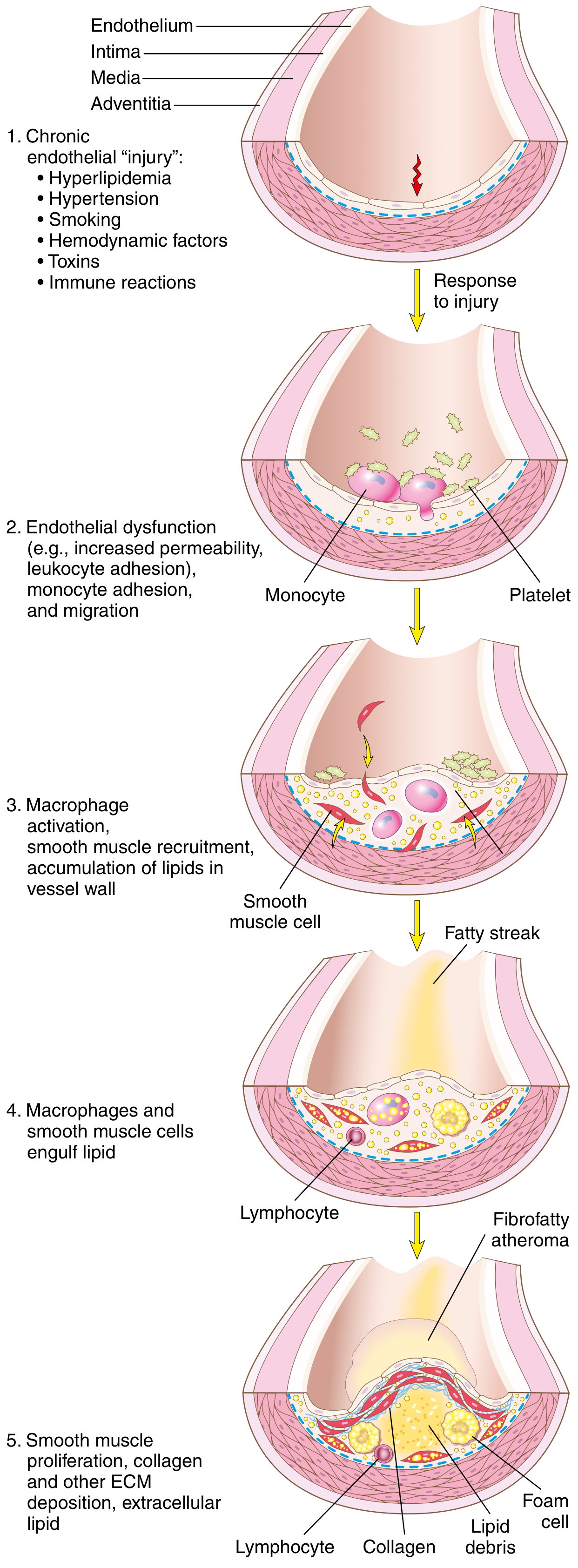
Step-by-Step Sequence:
Step 1 - Endothelial Injury & Dysfunction
- Endothelial cell (EC) injury is the cornerstone of atherosclerosis
- Early lesions begin at sites of intact but dysfunctional endothelium
- "Dysfunction" = defective vascular tone, impaired barrier function, reduced hemostatic regulation, and enhanced inflammation
- Two most important causes of EC dysfunction:
- Hemodynamic disturbances - Turbulent blood flow at ostia, branch points, and posterior abdominal aorta preferentially induces EC injury. Non-turbulent laminar flow induces "atheroprotective" genes - explaining why plaques form at specific anatomical sites
- Hypercholesterolemia - Chronic hyperlipidemia directly impairs ECs by increasing reactive oxygen species (ROS), which accelerate nitric oxide (NO) decay, dampening vasodilator activity
- Other triggers: inflammatory cytokines (TNF-α), cigarette smoke toxins, hypertension, immune reactions
Step 2 - Lipoprotein Accumulation & Oxidation
- With chronic hyperlipidemia, LDL enters and accumulates in the intima
- Oxidized LDL (ox-LDL) forms when LDL is oxidized by free radicals from inflammatory cells - this is particularly dangerous:
- Ox-LDL is toxic to ECs, SMCs, and macrophages
- It is taken up by macrophages via scavenger receptors (not the normal LDL receptor) - leading to foam cell formation since it cannot be degraded
- Stimulates release of growth factors, cytokines, and chemokines
- Recruits more monocytes - creating a vicious cycle
Step 3 - Monocyte Recruitment & Macrophage Activation
- Dysfunctional ECs express adhesion molecules (VCAM-1, ICAM-1, E-selectin) that capture monocytes and T lymphocytes from blood
- Monocytes migrate into the intima via chemokines (e.g., MCP-1) and differentiate into macrophages
- Macrophages engulf oxidized LDL via scavenger receptors → transform into foam cells (lipid-laden macrophages)
- Macrophages also sense cholesterol crystals and free fatty acids via the inflammasome → produce IL-1 → activates ECs, recruits more mononuclear cells
- Activated macrophages produce: ROS (enhance LDL oxidation), growth factors (drive SMC proliferation), and proteases (break down ECM)
Step 4 - Platelet Adhesion
- Exposed intimal components trigger platelet adhesion and activation
- Activated platelets release platelet-derived growth factor (PDGF) and other cytokines
Step 5 - T-Cell Recruitment & Inflammatory Amplification
- T lymphocytes enter the intima and release interferon-γ (IFN-γ) which:
- Activates macrophages further
- Activates ECs and SMCs
- Promotes inflammation (contributing to plaque instability)
Step 6 - Smooth Muscle Cell (SMC) Proliferation & Matrix Synthesis
- Growth factors (PDGF from platelets/macrophages, FGF, TGF-β) recruit SMCs from the media into the intima (phenotypic switch: contractile → synthetic)
- Intimal SMCs proliferate and synthesize ECM (especially collagen) - converting the fatty streak into a mature atheroma
- Collagen deposition forms and stabilizes the fibrous cap
- However, activated macrophages can also induce SMC apoptosis and ECM breakdown → unstable plaques
Step 7 - Lipid Accumulation (Extracellular)
- As foam cells die, lipids spill into the extracellular space → necrotic lipid core forms
- Cholesterol crystals become incorporated into the plaque
4. Morphology (Lesion Types)
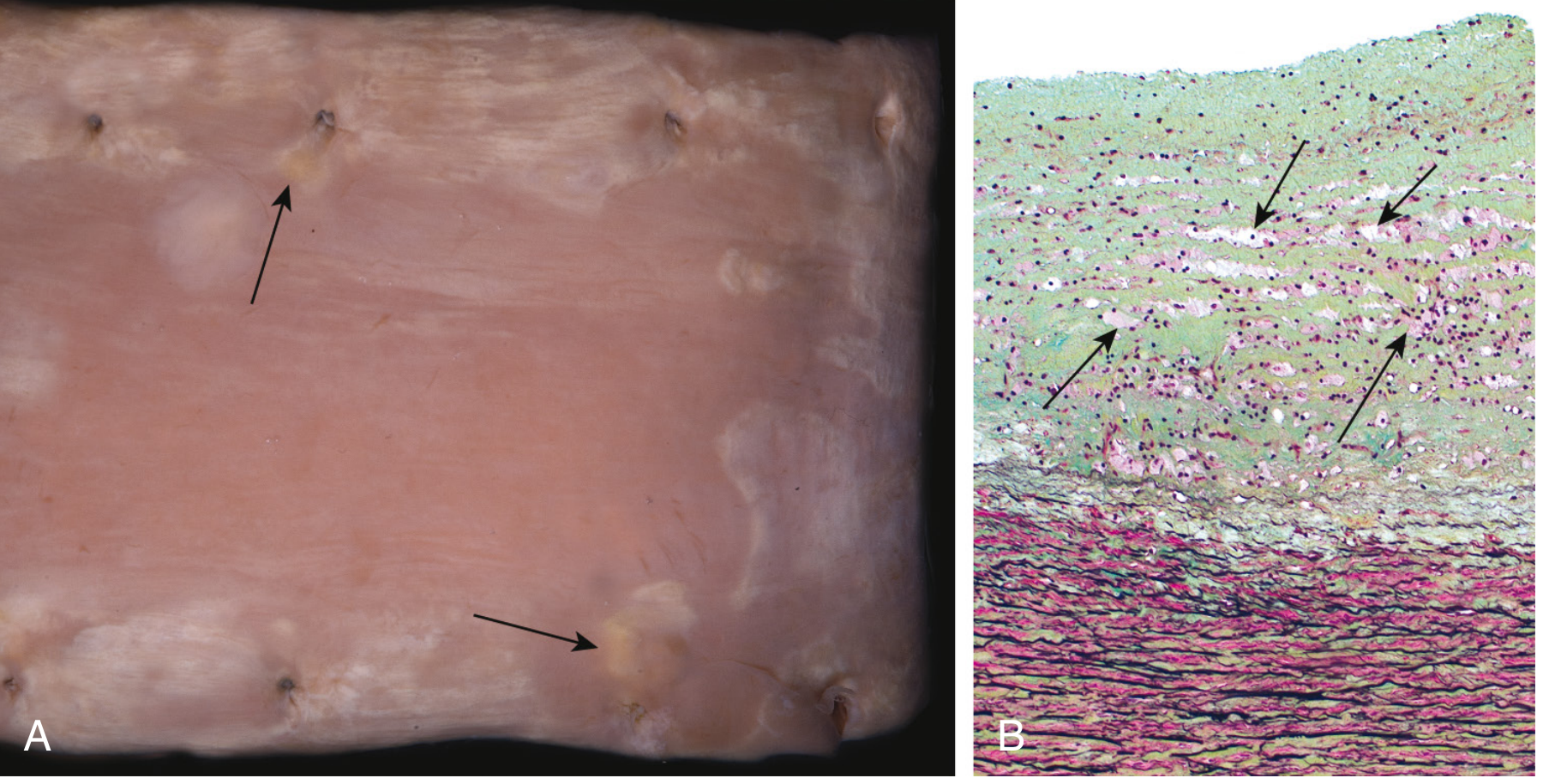
Stage 1 - Fatty Streak (Earliest Lesion)
- Gross: Minute, flat, yellow macules that coalesce into elongated lesions ≥1 cm long
- Microscopy: Intimal lipid-filled foam cells (macrophage-derived) - no significant flow disturbance
- Age: Can appear in the aorta of infants <1 year old; present in virtually ALL children >10 years, regardless of risk factors
- Significance: Not all progress to plaques - but coronary fatty streaks form during adolescence at the same sites that develop plaques later in life
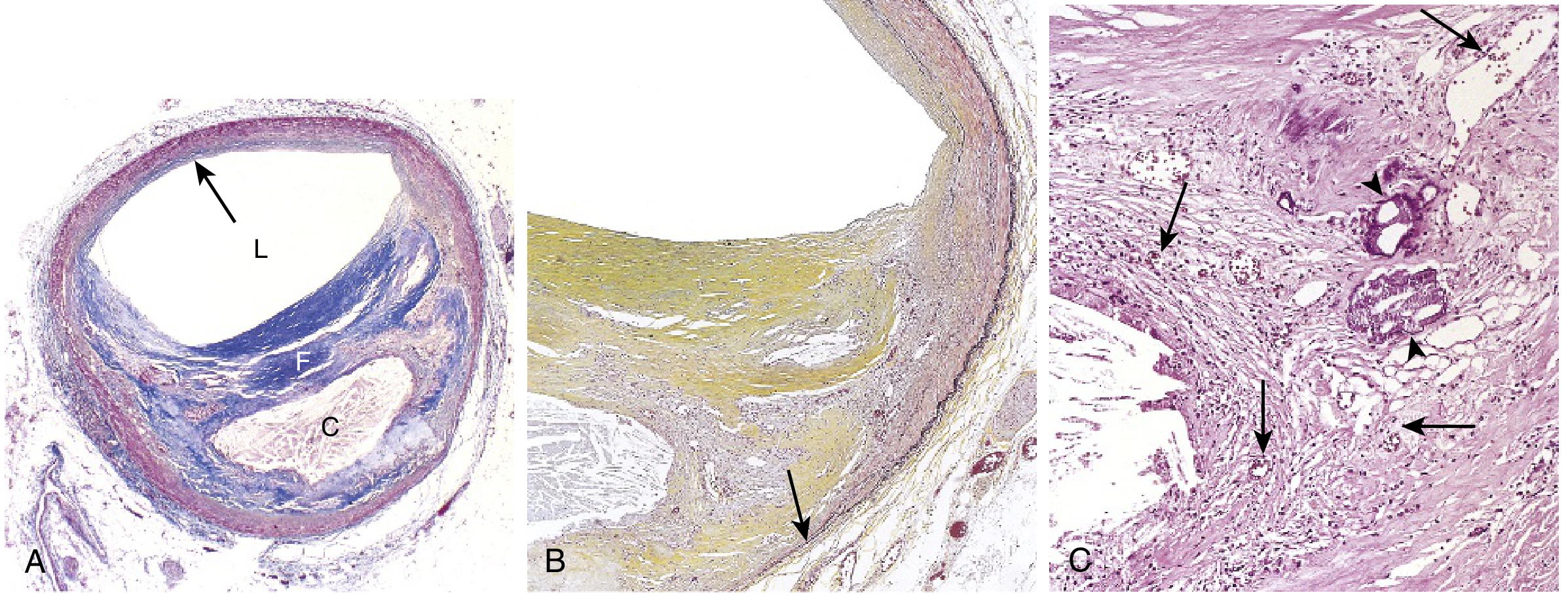
Stage 2 - Atherosclerotic Plaque (Atheromatous Plaque / Fibrous Plaque)
- Gross: White to yellow raised lesions, 0.3-1.5 cm in diameter (may coalesce); eccentric (involves only part of the vessel wall)
- Microscopy: Three key components:
- Fibrous cap - Smooth muscle cells, collagen (dense), proteoglycans, elastin; relatively few inflammatory cells
- Cellular zone (shoulder region) - Active area with macrophages, foam cells, T lymphocytes, SMCs; most prone to rupture
- Necrotic lipid core - Extracellular lipids, cholesterol crystals, necrotic debris, calcification; no collagen
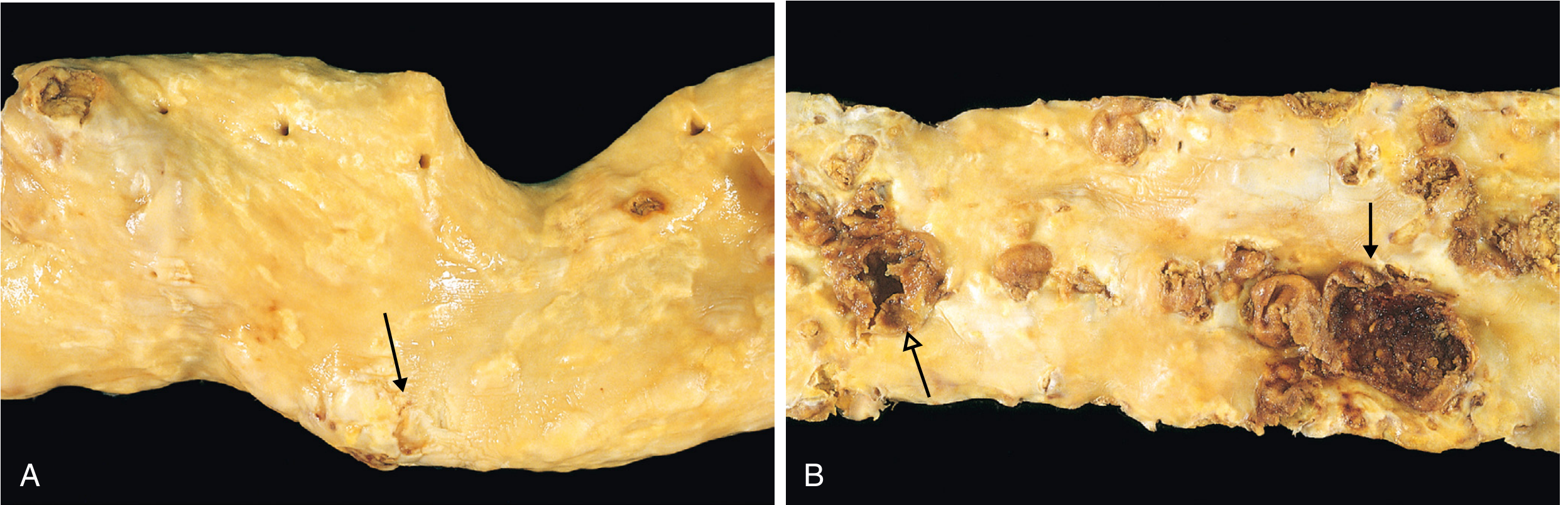
Distribution of Plaques (Descending Frequency):
- Infrarenal abdominal aorta (most common/severe)
- Coronary arteries
- Popliteal arteries
- Internal carotid arteries
- Circle of Willis
5. Stable vs. Vulnerable (Unstable) Plaque
This is the most clinically important distinction:
| Feature | Stable Plaque | Vulnerable (Unstable) Plaque |
|---|---|---|
| Fibrous cap | Thick, rich in collagen | Thin, poorly supported |
| Lipid core | Small | Large (>40% of plaque volume) |
| Inflammatory cells | Few | Abundant macrophages and T cells |
| SMCs | Abundant | Reduced (apoptotic) |
| Clinical behavior | Stable angina | ACS: unstable angina, STEMI, NSTEMI |
| Risk | Gradual stenosis | Rupture → thrombosis |
Plaque destabilization is driven by:
- Macrophage-derived metalloproteinases (MMPs) that digest collagen in the fibrous cap
- T cell IFN-γ inhibiting collagen synthesis by SMCs
- SMC apoptosis from inflammatory signals
6. Consequences of Atherosclerosis / Acute Plaque Change
Plaque changes fall into three categories:
1. Plaque Rupture / Erosion / Ulceration
- Exposes highly thrombogenic subendothelial components and lipid core
- Triggers thrombus formation - can cause acute coronary syndrome (unstable angina, STEMI, NSTEMI)
- Most ruptures occur at the shoulder region of the plaque (highest inflammatory cell density, thinnest fibrous cap)
2. Hemorrhage into Plaque
- Rupture of overlying fibrous cap or thin-walled neovessels within the plaque
- Sudden expansion of plaque volume → acute luminal narrowing
3. Atheroembolism
- Plaque rupture can discharge atherosclerotic debris (cholesterol crystals, etc.) into the bloodstream
- Causes microemboli downstream (e.g., "blue toe syndrome")
4. Aneurysm Formation
- Atherosclerosis-induced ischemia of the underlying media with loss of elastic tissue → wall weakening → aneurysmal dilation and rupture
- Most common: infrarenal abdominal aortic aneurysm (AAA)
Atherosclerotic Stenosis (Chronic)
- Critical stenosis: ~70% occlusion is the tipping point in coronary arteries
- At rest: adequate perfusion; with exertion: demand exceeds supply → stable angina
- Chronic arterial hypoperfusion causes: bowel ischemia, chronic IHD, ischemic encephalopathy, intermittent claudication
7. Major Clinical Consequences
| Artery Involved | Clinical Consequence |
|---|---|
| Coronary arteries | Myocardial infarction, stable angina, sudden cardiac death |
| Cerebral / carotid arteries | Ischemic stroke, TIA |
| Aorta | Aortic aneurysm (AAA), aortic dissection |
| Peripheral arteries (popliteal, iliac) | Peripheral arterial disease, intermittent claudication, gangrene |
| Renal arteries | Renovascular hypertension, renal failure |
| Mesenteric arteries | Bowel ischemia / infarction |
8. Key High-Yield Points for MBBS Exams
- Response to injury hypothesis (Ross) = the core concept - endothelial injury is the initiating event
- Foam cells = lipid-laden macrophages (and SMCs) formed via scavenger receptor uptake of ox-LDL
- Fatty streak = earliest visible lesion; present in virtually all children >10 years; does NOT obstruct flow
- Fibrous cap = collagen + SMCs; protects against plaque rupture
- Vulnerable plaque = thin cap, large lipid core, heavy inflammation - responsible for ACS
- Most severe atherosclerosis = infrarenal abdominal aorta > coronary > popliteal > internal carotid
- Critical stenosis = 70% in coronary arteries = stable angina threshold
- CRP = strong independent inflammatory marker of cardiovascular risk (not causative)
- Hyperhomocysteinemia - additional risk factor (caused by vit B12/B6/folate deficiency or inborn errors)
- Diabetes = 100-fold increased risk of lower limb gangrene
Summary Diagram of Pathogenesis (Memorization Framework)
Endothelial Injury (hyperlipidemia, HTN, smoking, turbulence)
↓
Endothelial Dysfunction (↑ permeability, ↑ leukocyte adhesion)
↓
LDL enters intima → Oxidized LDL (ox-LDL)
↓
Monocytes adhere (VCAM-1) → migrate → Macrophages
↓
Macrophages engulf ox-LDL via scavenger receptors → FOAM CELLS → FATTY STREAK
↓
Platelet adhesion → PDGF released
Macrophages → IL-1, TNF-α, ROS, MMPs
T cells → IFN-γ
↓
SMC migration (media → intima) + proliferation + collagen synthesis
↓
Fibrous cap forms over lipid core → ATHEROSCLEROTIC PLAQUE
↓
Plaque growth → stenosis OR plaque rupture → THROMBOSIS → ACS/Stroke
Robbins & Kumar Basic Pathology, 10e, pp. 314-321 | Robbins, Cotran & Kumar Pathologic Basis of Disease, 10e, pp. 466-478
This is a shared conversation. Sign in to Orris to start your own chat.