Hypertension pathogenesis
Reading File
search_medical_text
hypertension pathogenesis mechanisms pathophysiology
search_medical_text
RAAS sympathetic nervous system vascular remodeling hypertension
search_medical_image
hypertension pathogenesis diagram RAAS
GradeResponse
GradeResponse
GradeResponse
Hypertension Pathogenesis
Core Hemodynamic Framework
Arterial pressure is the product of two variables (Harrison's, p. 7634):
BP = Cardiac Output (CO) × Peripheral Vascular Resistance (PVR)
- CO is determined by stroke volume (myocardial contractility + vascular compartment size) and heart rate.
- PVR is governed by functional and structural changes in small arteries (lumen 100–400 μm) and arterioles.
Hypertension arises when one or both of these are chronically elevated, through a complex interplay of genetic, environmental, neurohumoral, and organ-level mechanisms.
Major Pathophysiological Mechanisms
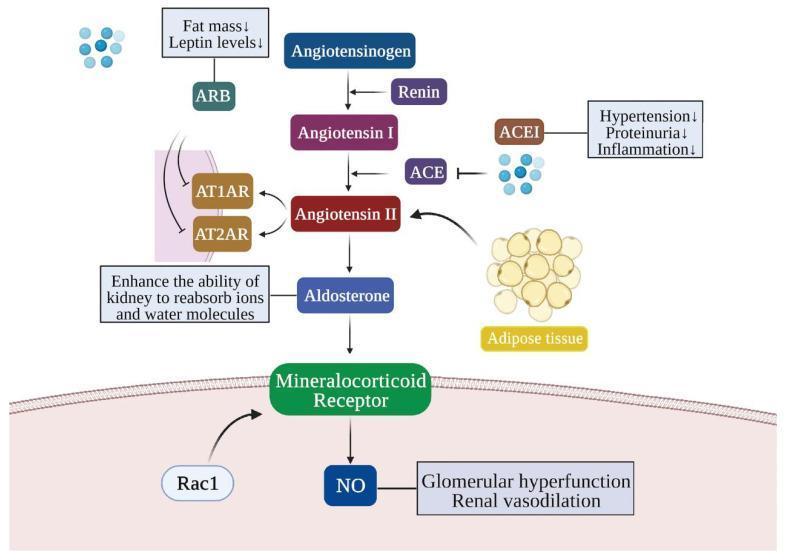
1. Renin-Angiotensin-Aldosterone System (RAAS)
The RAAS is a central driver of BP regulation and hypertension:
- Renin (released by juxtaglomerular cells in response to low renal perfusion, low Na⁺, or SNS activation) cleaves angiotensinogen → Angiotensin I
- ACE (in lung endothelium) converts Ang I → Angiotensin II (Ang II)
- Ang II acts on AT₁ receptors to cause:
- Potent vasoconstriction (↑ PVR)
- Aldosterone release → Na⁺ and water retention (↑ CO)
- Direct renal tubular Na⁺ reabsorption
- Vascular smooth muscle hypertrophy (structural remodeling)
- Central sympathetic activation
- Oxidative stress and endothelial dysfunction
2. Sympathetic Nervous System (SNS) Overactivity
- Increased efferent SNS tone elevates BP via:
- Increased heart rate and contractility (↑ CO)
- Vasoconstriction (↑ PVR)
- Renal vasoconstriction → renin release (activating RAAS)
- Na⁺ retention
- Triggers include stress, obesity (leptin-mediated), sleep apnea, and baroreceptor dysfunction
- Chronic SNS activation resets the baroreflex to a higher set point
3. Renal Mechanisms — Pressure-Natriuresis Dysfunction
The kidney normally adjusts Na⁺ excretion in proportion to perfusion pressure (Guyton's model). In hypertension:
- The pressure-natriuresis curve is shifted rightward — higher BP is required to achieve the same natriuresis
- Causes include reduced nephron number (Brenner hypothesis), intrarenal RAAS activation, and tubular Na⁺ transporter upregulation (NHE3, ENaC)
- Volume expansion → sustained ↑ CO → ↑ BP
4. Vascular Remodeling and Endothelial Dysfunction
| Feature | Effect |
|---|---|
| Reduced nitric oxide (NO) bioavailability | Impaired vasodilation, ↑ PVR |
| ↑ Reactive oxygen species (ROS) | NO scavenging, oxidative endothelial injury |
| Smooth muscle hypertrophy/hyperplasia | Permanent ↑ in wall:lumen ratio |
| ↑ Endothelin-1 | Vasoconstriction |
| Arterial stiffness (↓ elastin, ↑ collagen) | ↑ Pulse wave velocity, isolated systolic HTN |
Ang II, aldosterone, and chronic shear stress all contribute to these structural changes, which make hypertension self-perpetuating.
5. Genetic and Polygenic Contributions
- Hypertension is highly heritable (~30–50%)
- Monogenic forms (rare): Liddle syndrome (ENaC gain-of-function), glucocorticoid-remediable aldosteronism, Gordon syndrome (WNK kinase mutations)
- Common/polygenic: hundreds of SNPs each with small effect, involving Na⁺ transport, RAAS components, adrenergic receptors, and endothelial function
- As noted in the management guidelines (GLGCA, p. 17), "the contribution of these factors may differ among males and females"
6. Salt Sensitivity and Volume Dysregulation
- ~50% of hypertensives are salt-sensitive (BP rises with Na⁺ load)
- More prevalent in Black populations, elderly, CKD, and diabetics
- Mechanisms: blunted renal dopamine response, RAAS over-activation, aldosterone excess, reduced renal medullary prostaglandins
7. Neuroinflammation and Immune Activation
- T lymphocytes (particularly Th17 cells) infiltrate the kidney and vasculature in hypertension
- Ang II drives T-cell activation → release of IL-17, TNF-α
- These cytokines promote renal Na⁺ retention and vascular inflammation
- An emerging mechanism explaining why anti-inflammatory pathways can lower BP experimentally
8. Obesity and Metabolic Factors
- Adipose tissue is a source of angiotensinogen (direct RAAS input)
- Leptin hypersecretion activates the SNS (renal and adrenal)
- Insulin resistance → hyperinsulinemia → Na⁺ retention, SNS activation
- Obstructive sleep apnea causes intermittent hypoxia → chemoreceptor activation → chronic SNS elevation
Integrated Model
Genetic predisposition + Environmental factors (Na⁺, obesity, stress)
↓
↑ RAAS + ↑ SNS + Renal Na⁺ retention
↓
↑ CO and/or ↑ PVR → Elevated BP
↓
Vascular remodeling + Endothelial dysfunction
↓
BP elevation becomes structural and self-sustaining
(Management of Elevated Blood Pressure and Hypertension, p. 17; Harrison's, p. 7634)
Secondary Hypertension — Distinct Pathogenic Pathways
| Cause | Mechanism |
|---|---|
| Primary hyperaldosteronism | Autonomous aldosterone → Na⁺/water retention |
| Renal artery stenosis | ↓ Renal perfusion → ↑ renin → RAAS activation |
| Pheochromocytoma | Catecholamine excess → ↑ CO and ↑ PVR |
| Cushing's syndrome | Glucocorticoid-mediated Na⁺ retention, ↑ vascular reactivity |
| CKD | ↓ Nephron mass, volume overload, RAAS activation |
| Hypothyroidism | ↓ CO but ↑ PVR (diastolic HTN) |
Understanding these overlapping mechanisms is the basis for rational antihypertensive therapy — diuretics (volume), ACEi/ARBs (RAAS), beta-blockers (SNS/CO), CCBs (PVR), and aldosterone antagonists each target distinct nodes in the pathogenic cascade.
This is a shared conversation. Sign in to Orris to start your own chat.