Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Searching Images
acute respiratory distress syndrome pathophysiology mechanism
Finding Sources
Loading Image
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive mechanistic content from multiple authoritative sources. Let me compile the full answer.
Acute Respiratory Distress Syndrome (ARDS) — Mechanism
Definition & Berlin Criteria
ARDS is a syndrome of acute hypoxemic respiratory failure characterised pathologically by diffuse alveolar damage (DAD). The 2012 Berlin Definition requires all four criteria:
| Criterion | Threshold |
|---|---|
| Timing | Onset within 7 days of a known clinical insult |
| Imaging | Bilateral opacities on CXR/CT, not explained by effusion, atelectasis, or cardiac failure |
| Origin | Not attributable to cardiac failure or fluid overload |
| Oxygenation (P/F ratio) | Mild: 200–300 mmHg · Moderate: 100–200 mmHg · Severe: <100 mmHg (all with PEEP ≥5 cmH₂O) |
Common Triggers
ARDS arises from direct (pulmonary) or indirect (extrapulmonary) insults to the alveolar-capillary membrane:
- Direct: pneumonia (35–45%), aspiration, inhalation injury, pulmonary contusion
- Indirect: sepsis (30–35%), severe pancreatitis, multiple trauma, massive transfusion, COVID-19
Core Pathophysiology
The central event is injury to both the alveolar epithelium and the vascular endothelium, driven by uncontrolled inflammation. This unfolds in overlapping phases:
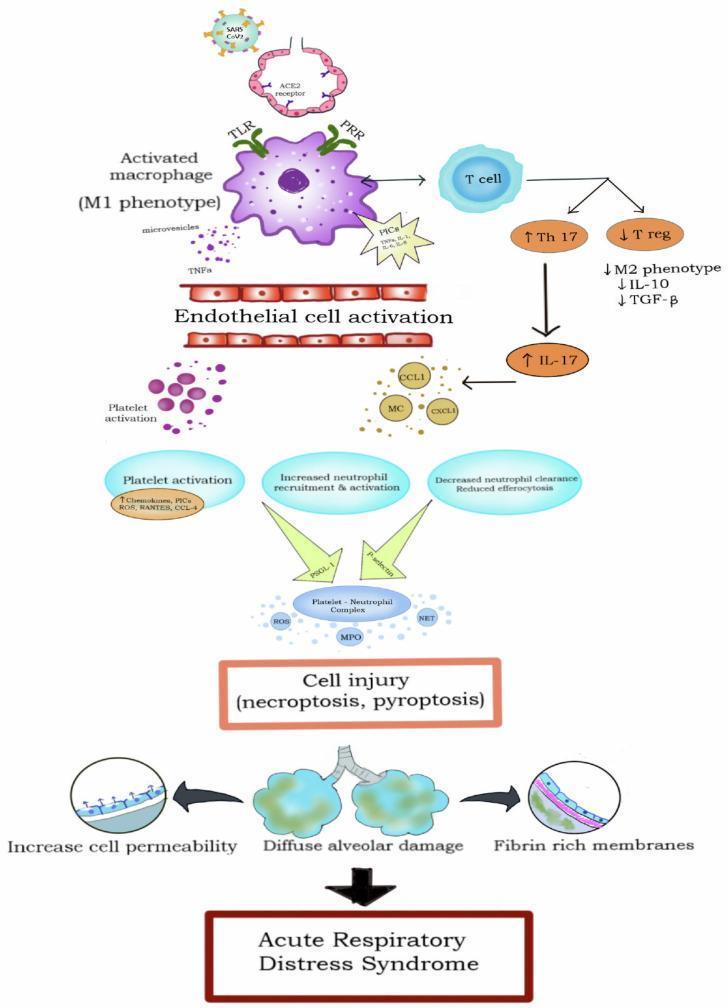
Phase 1 — Initiating Insult & Innate Immune Activation
- A systemic or pulmonary insult activates alveolar macrophages (M1 phenotype) via pattern-recognition receptors (TLR/PRR).
- Macrophages release IL-1, TNF-α, IL-6, IL-8, and other proinflammatory cytokines.
- These cytokines activate pulmonary endothelial cells, upregulating adhesion molecules (ICAM-1, P-selectin) and triggering neutrophil sequestration within pulmonary capillaries.
Phase 2 — Neutrophil-Mediated Alveolar Injury
Neutrophils are central effectors. Histology in early ARDS shows increased neutrophils in capillaries, interstitium, and alveolar spaces. Activated neutrophils release:
- Reactive oxygen species (ROS) — oxidative membrane damage
- Proteases (elastase, MMPs) — degrade basement membrane and extracellular matrix
- Neutrophil extracellular traps (NETs) — promote further inflammation and coagulation
- Platelet-activating factor — amplifies vascular permeability
The combined assault on the type I pneumocytes (gas exchange cells) and the capillary endothelium destroys the alveolar-capillary barrier.
Phase 3 — Loss of Barrier Integrity & Alveolar Flooding
Disruption of the alveolar-capillary membrane causes:
- Increased vascular permeability → protein-rich fluid floods the alveolar space (noncardiogenic pulmonary oedema)
- Inactivation of surfactant (by phospholipase A₂ and edema fluid dilution) → alveolar collapse and reduced compliance
- Hyaline membrane formation — fibrin-rich exudate mixed with necrotic epithelial cell debris lines the alveolar ducts (the pathological hallmark)
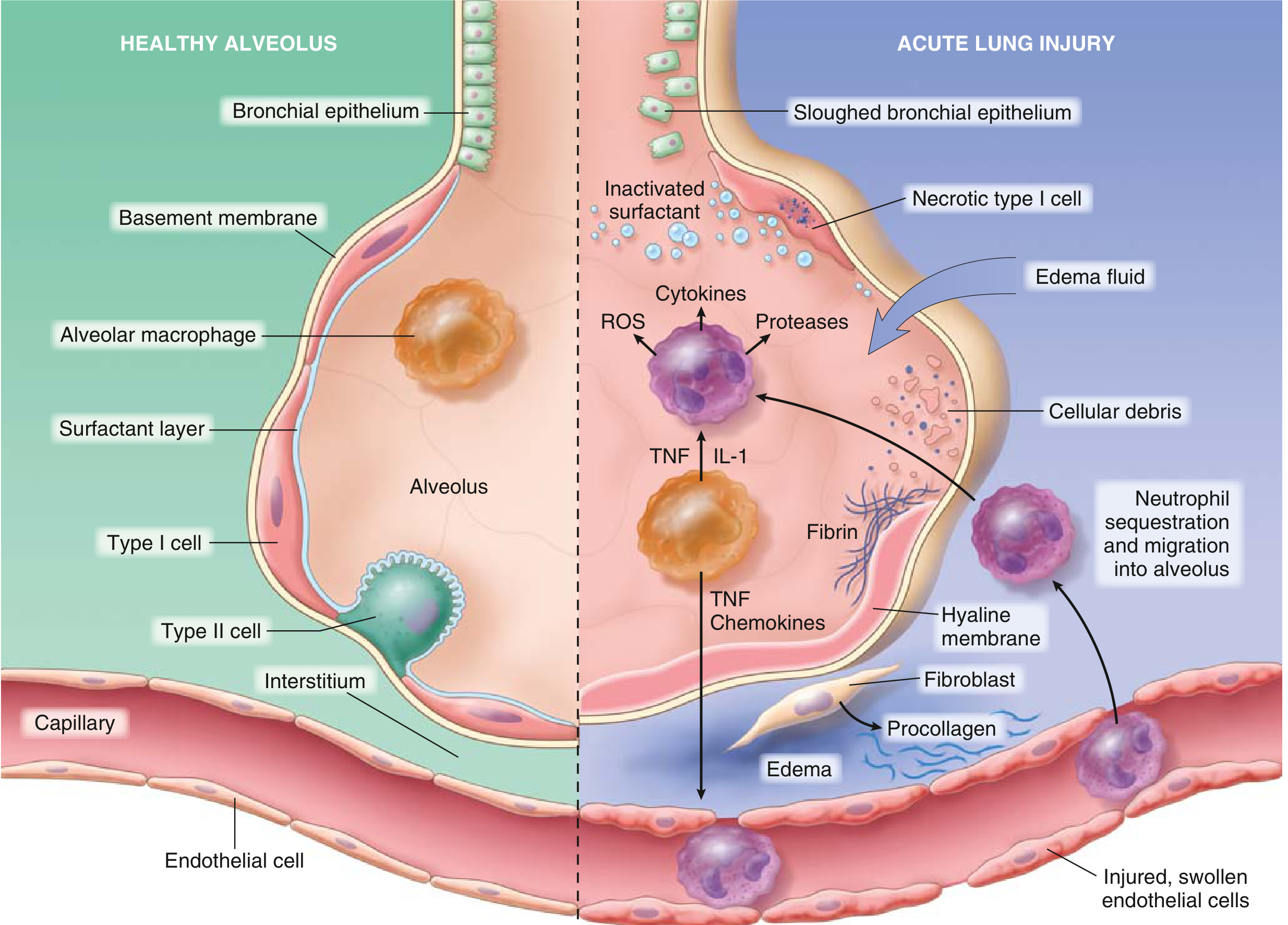
Phase 4 — Ventilation-Perfusion Mismatch & Hypoxaemia
- Fluid-filled and collapsed alveoli receive blood flow but cannot oxygenate it → intrapulmonary shunt (V/Q = 0)
- Reduced functional residual capacity (FRC) and decreased lung compliance (stiff lung)
- Hypoxic vasoconstriction is overwhelmed, worsening shunt
- Result: refractory hypoxaemia unresponsive to supplemental O₂ alone
Cellular & Molecular Cascade (Summary Diagram)
Phases of Histological Evolution
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Alveolar oedema, neutrophil influx, hyaline membranes, necrosis of type I pneumocytes |
| Fibroproliferative | Days 7–21 | Type II pneumocyte proliferation (attempted repair), fibroblast infiltration, early collagen deposition |
| Fibrotic | >3 weeks | Alveolar septal thickening, interstitial fibrosis, loss of normal architecture |
Complete resolution is uncommon. More often, the fibrin-rich exudate undergoes organisation leading to fibrosis.
Gross/Microscopic Morphology
- Gross: Dark red, firm, heavy, airless ("liver-like") lungs
- Micro: Capillary congestion, interstitial and intra-alveolar oedema and haemorrhage, neutrophilic infiltrate, hyaline membranes lining alveolar ducts (hallmark), necrotic type I cells
- Resolving phase: Proliferating type II pneumocytes, fibroblasts, collagen — alveolar septal thickening
Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
Because the lung is heterogeneously injured (dependent zones are consolidated; non-dependent zones remain aerated — the "baby lung" concept), mechanical ventilation can amplify injury through:
- Volutrauma/barotrauma: overdistension of aerated alveoli at high tidal volumes or pressures
- Atelectrauma: repetitive opening/collapse of unstable alveoli
- Biotrauma: mechanical stretch releases additional cytokines (IL-1β, IL-6, TNF-α) that spill into systemic circulation, contributing to multi-organ failure
This is why lung-protective ventilation (6 mL/kg tidal volume, plateau pressure ≤30 cmH₂O) is the cornerstone of management.
Key Physiological Consequences
| Consequence | Mechanism |
|---|---|
| Refractory hypoxaemia | Intrapulmonary shunt, V/Q mismatch |
| Reduced compliance | Surfactant loss, alveolar flooding, atelectasis |
| Reduced FRC | Alveolar collapse, oedema |
| Pulmonary hypertension | Hypoxic vasoconstriction, endothelial injury, microthrombi |
| Multi-organ failure | Biotrauma, systemic cytokine release |
Sources: Robbins & Kumar Basic Pathology, p. 444–445 · Barash Clinical Anaesthesia 9e, p. 4842–4843 · Murray & Nadel's Textbook of Respiratory Medicine · Goldman-Cecil Medicine, p. 1066–1067 · Sabiston Textbook of Surgery, p. 940
This is a shared conversation. Sign in to Orris to start your own chat.