Hepatitis
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive, authoritative content from Robbins Pathology and Yamada's Gastroenterology. Let me compose a thorough response.
Viral Hepatitis - Pathophysiology & Mechanisms
Overview
Five hepatotropic viruses - A, B, C, D, and E - account for 90-95% of all acute viral hepatitis cases. They are a genetically diverse group linked by their tropism for the liver (their primary site of replication), not by molecular structure. Hepatitis B, C, D, and in some circumstances E can progress to chronic hepatitis; HAV is always self-limited.
Hepatitis A Virus (HAV)
Virology: A small, nonenveloped, positive-strand RNA picornavirus in the genus Hepatovirus, 27 nm in diameter (icosahedral capsid). Discovered in 1973.
Receptor: HAVcr-1 (TIM-1), a glycoprotein on hepatocytes.
Pathogenesis:
- HAV is not cytopathic - it does not directly destroy cells
- Hepatocellular injury is inflicted by cytotoxic T lymphocytes (CTL) and NK cells that recognize and kill infected hepatocytes
- Spread is fecal-oral; the virus sheds in stool for 2-3 weeks before and 1 week after jaundice onset
- Viremia is transient, making bloodborne transmission rare
Outcome: Always self-limited. No chronic hepatitis, no carrier state. Case fatality rate < 0.5% in most populations, but severe in the elderly and those with pre-existing liver disease.
Hepatitis B Virus (HBV)
Virology: A small, enveloped, partially double-stranded DNA virus (hepadnavirus). The genome encodes four proteins:
| Protein | Function |
|---|---|
| HBsAg (surface antigen) | Envelope protein; released in excess as non-infectious particles; basis for vaccination |
| HBcAg (core antigen) | Nucleocapsid protein; assists viral assembly |
| HBeAg | Pre-core/core polypeptide; marker of active replication & infectivity |
| HBx protein | Transcriptional activator of viral and host genes; implicated in HCC pathogenesis |
| HBV polymerase | Has both DNA polymerase and reverse transcriptase activity |
Replication cycle:
- Large HBsAg binds to the bile salt transporter NTCP (sodium taurocholate cotransporting polypeptide) on hepatocytes
- Viral genome enters the nucleus and forms covalently closed circular DNA (cccDNA) - the transcriptional template
- Replication proceeds through reverse transcription via an RNA intermediate (like retroviruses)
- cccDNA persists in the nucleus as a minichromosome, making complete cure difficult
Immune Pathogenesis:
- High-level viral replication can be directly cytopathic, but most hepatocyte injury is immune-mediated
- CD8+ cytotoxic T cells attack infected hepatocytes
- A strong CD4+ and CD8+ IFN-γ response = resolution of acute infection
- A weak or dysregulated immune response = chronic infection and progressive liver damage
Serologic markers and their meaning:
| Marker | Meaning |
|---|---|
| HBsAg | Active infection (acute or chronic) |
| Anti-HBs | Recovery or vaccine immunity |
| IgM anti-HBc | Recent/acute infection ("window period" marker) |
| HBeAg | Active viral replication, high infectivity |
| Anti-HBe | Waning acute infection or mutation preventing HBeAg production |
| HBV DNA | Direct measure of viral load |
Chronicity: ~2/3 of acute infections are mild or subclinical. Chronic infection occurs in ~5-10% of adults but rises to >90% in neonates (immature immune system). Chronic HBV significantly raises the risk of cirrhosis and hepatocellular carcinoma (HCC) - partly via HBx-mediated transcriptional activation.
Hepatitis C Virus (HCV)
Virology: A small, enveloped, single-stranded positive-sense RNA virus in the family Flaviviridae. Discovered in 1989.
Key structural proteins:
- E1 & E2 - envelope glycoproteins; E2 is the primary target for neutralizing antibodies
- NS3/4A - serine protease (target of current antivirals)
- NS5A - replication complex protein (target of DAAs)
- NS5B - RNA-dependent RNA polymerase (target of sofosbuvir)
Pathogenesis and immune evasion - the key to HCV's success:
- High mutation rate: The NS5B RNA polymerase has low fidelity, generating vast genetic diversity. This creates 7 genotypes and numerous subtypes
- Quasispecies: Within each infected person, multiple genetic variants (quasispecies) emerge over time
- Antibody escape: Neutralizing antibodies target E2 - but emerging variants with altered E2 epitopes evade them
- Innate immune sabotage: NS3/NS4A protease cleaves MAVS and TRIF - adaptor proteins critical for interferon signaling - blunting the host antiviral response
Chronicity: Spontaneous clearance occurs in only ~15-20% of people. Chronic infection develops in 80-90%. Of those with chronic HCV:
- ~20% progress to cirrhosis over 20-30 years
- Those with cirrhosis have substantial risk of HCC
Progression is accelerated by: older age, male sex, alcohol use, immunosuppression, HBV/HIV co-infection, obesity, type 2 diabetes, and metabolic syndrome.
Hepatitis D Virus (HDV)
Virology: A unique satellite virus - it is a defective RNA virus that can only replicate in cells simultaneously infected with HBV. It uses HBsAg as its own outer envelope.
Two patterns of infection:
| Pattern | Description | Outcome |
|---|---|---|
| Co-infection | Simultaneous HDV + HBV infection | Usually self-limited; fulminant hepatitis possible |
| Superinfection | HDV infects a chronic HBV carrier | >80% develop chronic HDV; severe exacerbation common |
Pathogenesis: HDV RNA is detectable before symptoms. In superinfection with chronic HDV there are two phases - an acute phase with active HDV replication and suppression of HBV replication (high transaminases), and a chronic phase where HDV replication falls and HBV replication rebounds.
Key point: HBV vaccination prevents HDV by eliminating the host requirement.
Hepatitis E Virus (HEV)
Virology: An unenveloped, positive-stranded RNA virus in the genus Orthohepevirus (family Hepeviridae). Genome is 7.3 kb with 4 open reading frames encoding viral protease and RNA polymerase.
Transmission: Fecal-oral, waterborne (zoonosis - reservoirs include pigs, monkeys, dogs, cats).
Pathogenesis:
- HEV is not cytopathic - hepatic damage stems from the host immune response to infected cells
- Incubation period: 4-5 weeks
- Virions shed in stool during acute illness
- HEV RNA and virions detectable in stool and serum before symptoms
- IgM anti-HEV appears simultaneously with clinical illness, then replaced by persistent IgG
Special populations:
- Pregnant women: Mortality rate approaching 20% - the mechanism is thought to involve immune dysregulation of pregnancy
- Immunocompromised patients (AIDS, transplant recipients): Chronic HEV infection can occur
- Immunocompetent patients: Always self-limited, no chronic carrier state
Common Pathologic Mechanisms Across All Hepatitis Viruses
- Direct cytopathic injury - high-level viral replication (mainly HBV at peak viremia)
- T cell-mediated hepatocyte killing - the dominant mechanism for HAV, HBV, and HCV; CD8+ CTLs recognize MHC-I-presented viral peptides and destroy infected hepatocytes
- Immune complex-mediated extrahepatic disease - particularly HBV (glomerulonephritis, polyarteritis nodosa)
- Fibrogenesis and cirrhosis - chronic inflammation activates hepatic stellate cells (Ito cells) → collagen deposition → portal fibrosis → cirrhosis
- Hepatocellular carcinoma - HBV via HBx protein (transcriptional activation, genomic integration); HCV via chronic inflammation and oxidative stress
Summary Comparison Table
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Virus family | Picornaviridae | Hepadnaviridae | Flaviviridae | Satellite (deltaviridae) | Hepeviridae |
| Genome | +ssRNA | Partial dsDNA | +ssRNA | -ssRNA | +ssRNA |
| Transmission | Fecal-oral | Parenteral/sexual/vertical | Parenteral | Parenteral (requires HBV) | Fecal-oral |
| Chronicity | Never | Yes (~5-10% adults) | Yes (~80-90%) | Yes (>80% superinfection) | Rarely (immunocompromised) |
| Cytopathic? | No (T cell mediated) | Partial | No (immune mediated) | No | No (host immune response) |
| HCC risk | No | Yes (via HBx) | Yes (via cirrhosis) | Yes (additive to HBV) | No |
| Vaccine available | Yes | Yes | No | Prevented by HBV vaccine | Limited (China only) |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, 10e; Yamada's Textbook of Gastroenterology, 7e
Portal hypertension
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive breakdown of portal hypertension from pathophysiology to clinical complications.
Portal Hypertension
Definition and Measurement
Portal hypertension is defined as elevation of the hepatic venous pressure gradient (HVPG) above 5 mmHg. HVPG is the gold standard measurement - it represents the perfusion pressure of the liver with portal blood (measured by hepatic vein catheterization).
Key HVPG thresholds:
- >5 mmHg - portal hypertension (by definition)
- ≥6 mmHg - clinically measurable elevation
- ≥10 mmHg - clinically significant portal hypertension (CSPH); substantially increased decompensation risk
- ≥12 mmHg - variceal hemorrhage risk begins; target for TIPS reduction
Normal portal pressure is 5-10 mmHg. Once HVPG reaches ≥10 mmHg, the risk of decompensation (variceal bleeding, ascites, encephalopathy) rises sharply, and median survival after decompensation is less than 2 years.
Anatomy of the Portal Venous System
The portal vein carries blood from the entire GI tract (except upper esophagus and distal rectum), spleen, pancreas, and gallbladder into the liver. The superior mesenteric vein + splenic vein merge behind the neck of the pancreas to form the portal vein. The portal vein and hepatic artery drain into hepatic sinusoids, which drain into the hepatic veins and then the inferior vena cava (IVC).
The liver receives ~25-30% of cardiac output. Portal venous blood accounts for 75% of hepatic blood flow and ~50% of oxygen delivery. Sinusoids are highly permeable (fenestrated endothelium, no basement membrane), with the space of Disse containing Kupffer cells and hepatic stellate cells (HSCs) that regulate hepatic hemodynamics.
Pathophysiology
Portal venous pressure follows Ohm's law:
P = Q × R (pressure = flow × resistance)Therefore, portal hypertension results from:
- Increased resistance to portal blood flow
- Increased portal blood inflow (hyperdynamic circulation)
- In advanced disease: neurohumoral activation and systemic inflammation
Mechanism 1 - Increased Intrahepatic Resistance
In cirrhosis, two components raise intrahepatic resistance:
A. Structural (passive/fixed) component:
- Progressive collagen deposition and nodule formation distort sinusoidal architecture
- Hepatic stellate cells (HSCs) become activated into myofibroblasts and develop contractile properties
- This architectural distortion reduces the cross-sectional area of sinusoidal channels
B. Dynamic (functional/reversible) component (~30-40% of resistance):
This is therapeutically targetable:
- Nitric oxide (NO) deficiency: Despite excess NO production in the systemic/splanchnic circulation, intrahepatic eNOS activity is reduced in cirrhosis - the opposite of what occurs systemically. This leads to intrahepatic vasoconstriction
- Endothelin-1 (ET-1) excess: ET-1 binds ET-A receptors on HSCs → vasoconstriction
- Activated HSCs behave as pericytes - they contract around sinusoids and amplify resistance
- Imbalance between vasodilators (NO, prostacyclin) and vasoconstrictors (ET-1, angiotensin II, sympathetic tone) favors constriction in the portal bed
Mechanism 2 - Increased Portal Blood Inflow (Hyperdynamic Circulation)
As portal pressure rises, vasodilator substances (especially NO, prostacyclin, glucagon, substance P, endocannabinoids) produced in excess spill into the systemic/splanchnic circulation, causing:
- Splanchnic arterial vasodilation - markedly reduced splanchnic vascular resistance
- Increased splanchnic blood flow → augments portal venous inflow
- Systemic hyperdynamic state: high cardiac output, low systemic vascular resistance, low mean arterial pressure
This is a vicious cycle - the attempt to decompress the portal system by forming collaterals paradoxically sustains and amplifies it.
Mechanism 3 - Neurohumoral Activation and Systemic Inflammation
Splanchnic vasodilation reduces effective arterial blood volume (despite overall volume expansion). This triggers compensatory systems:
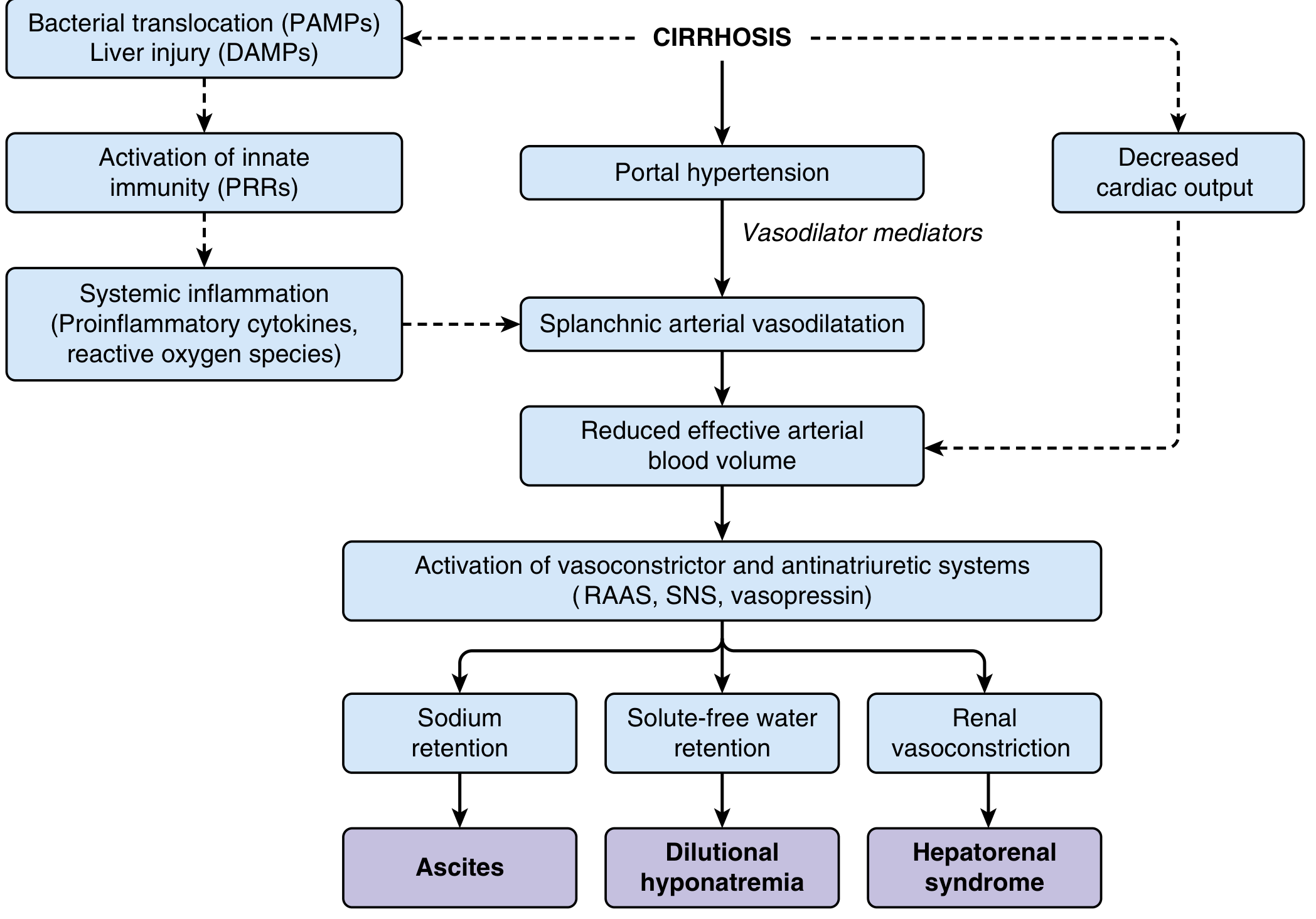
As shown above:
- RAAS activation → aldosterone → renal sodium and water retention → ascites
- Sympathetic nervous system (SNS) activation → renal vasoconstriction
- ADH (vasopressin) release → solute-free water retention → dilutional hyponatremia
- Bacterial translocation → gut flora traverse the intestine into mesenteric lymph nodes → PAMPs enter the circulation → pattern recognition receptors (PRRs) activate innate immunity → proinflammatory cytokines and ROS → further impair circulatory function
- In advanced disease: decreased cardiac output (cirrhotic cardiomyopathy) compounds effective hypovolemia
Classification by Anatomical Site
Intrahepatic causes account for >95% of all portal hypertension.
| Category | Subtype | Examples |
|---|---|---|
| Prehepatic | - | Portal vein thrombosis (PVT), splenic vein thrombosis, Banti's syndrome (massive splenomegaly) |
| Intrahepatic | Presinusoidal | Schistosomiasis, congenital hepatic fibrosis, primary biliary cholangitis (early) |
| Sinusoidal | Cirrhosis (all causes), alcoholic hepatitis | |
| Postsinusoidal | Sinusoidal obstruction syndrome (veno-occlusive disease) | |
| Posthepatic | - | Budd-Chiari syndrome (BCS), IVC webs, constrictive pericarditis, restrictive cardiomyopathy, right heart failure |
Clinical Complications
1. Gastroesophageal Varices
As portal pressure rises above the critical 12 mmHg threshold, portosystemic collaterals dilate. The gastroesophageal junction (GEJ) is the most clinically dangerous site because:
- The palisade zone (2-3 cm above the GEJ) is where portal and azygos venous systems communicate
- Incompetent valves in perforating veins allow retrograde flow, dilating intrinsic veins
- Variceal rupture in this zone causes massive upper GI hemorrhage
Variceal hemorrhage mortality: ~15-25% per episode. Rebleeding risk is 60-70% without prophylaxis.
2. Ascites
Mechanism: splanchnic vasodilation → reduced effective arterial volume → RAAS/SNS/ADH activation → renal sodium and water retention → fluid accumulates in the peritoneal cavity (hydrostatic pressure + reduced oncotic pressure from low albumin).
Diagnosed/categorized by the Serum-Ascites Albumin Gradient (SAAG):
- SAAG ≥1.1 g/dL (high gradient) = portal hypertension-related (cirrhosis, BCS, cardiac ascites, portal vein thrombosis)
- SAAG <1.1 g/dL (low gradient) = non-portal hypertension cause (malignancy, tuberculosis, pancreatitis)
3. Spontaneous Bacterial Peritonitis (SBP)
- Occurs in up to 30% of hospitalized cirrhotic patients with ascites
- Mechanism: bacterial translocation from gut (E. coli and other Gram-negatives most common)
- Diagnosis: ascitic fluid neutrophil count >250/μL
- Treatment: IV 3rd-generation cephalosporin × 5 days + IV albumin (1.5 g/kg day 1, 1.0 g/kg day 3) to prevent HRS
- Quinolone prophylaxis recommended after a first episode
4. Hepatorenal Syndrome (HRS)
Functional renal failure in ~10% of advanced cirrhosis or acute liver failure patients. No intrinsic renal pathology - it is driven by extreme renal vasoconstriction secondary to splanchnic vasodilation and RAAS/SNS activation.
Two patterns:
- HRS-AKI (formerly Type 1): Rapid progressive renal impairment (creatinine rise ≥0.3 mg/dL in 48h); high short-term mortality
- HRS-CKD (formerly Type 2): Stable, moderate renal impairment; better short-term outlook
Treatment: vasoconstrictors (terlipressin - preferred; norepinephrine in ICU; midodrine + octreotide as 3rd line) + IV albumin. Best treatment: liver transplantation.
5. Hepatic Encephalopathy (HE)
Neuropsychiatric complication from failure of the cirrhotic liver (and portosystemic shunting) to clear gut-derived neurotoxins, primarily ammonia. Ammonia metabolism by astrocytes produces glutamine, causing astrocyte swelling (cerebral edema). Additional contributors include manganese deposition in basal ganglia and neuroinflammation.
6. Splenomegaly and Hypersplenism
Congestion of the splenic vein → splenomegaly → sequestration of blood cells → thrombocytopenia, leukopenia, and anemia (hypersplenism). Thrombocytopenia also results from reduced thrombopoietin production by the diseased liver.
7. Hepatopulmonary Syndrome / Portopulmonary Hypertension
- Hepatopulmonary syndrome: Intrapulmonary vascular dilation → V/Q mismatch → hypoxemia. Characteristic: platypnea-orthodeoxia (desaturation on standing, relief on lying)
- Portopulmonary hypertension: Elevated mean pulmonary arterial pressure (>25 mmHg) due to portal hypertension-induced mediators reaching the pulmonary vasculature
Summary: The Dual-Hit Model
CIRRHOSIS
│
├─► Architectural distortion + HSC activation
│ → ↑ Intrahepatic resistance (structural + dynamic)
│ → ↑ Portal pressure
│
└─► Portal pressure → Splanchnic vasodilation (NO, prostacyclin)
→ ↑ Portal inflow (hyperdynamic circulation)
→ ↓ Effective arterial volume
→ RAAS / SNS / ADH activation
↓ ↓ ↓
Ascites Dilutional HypoNa HRS
Sources: Harrison's Principles of Internal Medicine 22e; Yamada's Textbook of Gastroenterology 7e; Sleisenger & Fordtran's Gastrointestinal and Liver Disease
This is a shared conversation. Sign in to Orris to start your own chat.