Aniridia
Reading File
Finding Sources
Finding Sources
Searching PubMed
aniridia PAX6 genetics management
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading Article
PMID: 40892309
I now have comprehensive information from multiple authoritative sources. Here is the full structured review:
Aniridia
Definition
Aniridia is a rare, bilateral panocular developmental disorder characterized by partial or complete absence of the iris. Despite the name, truly total absence of iris tissue is uncommon - gonioscopy almost always reveals a hypoplastic or rudimentary iris frill at the angle. It is best understood as a disorder of the master eye developmental gene PAX6, causing widespread ocular dysgenesis well beyond the iris alone.
- Kanski's Clinical Ophthalmology, 10th ed., p. 420
- The Wills Eye Manual, p. 537
Genetics
| Type | Frequency | Inheritance | PAX6 | Systemic Risk |
|---|---|---|---|---|
| Autosomal dominant (familial) | ~2/3 of cases | AD | PAX6 mutation | None |
| Sporadic (including WAGR) | ~1/3 of cases | New mutation | PAX6 deletion | Wilms tumor ~30% |
| Gillespie syndrome | ~1% | AR | NOT PAX6 | Cerebellar ataxia, learning disability |
Key molecular point: PAX6 lies adjacent to the WT1 gene on chromosome 11p13. When a larger chromosomal deletion encompasses both, the result is WAGR syndrome (Wilms tumor, Aniridia, Genital abnormalities, intellectual Retardation - previously called Miller syndrome). Autosomal dominant aniridia shows complete penetrance but variable expressivity (severity differs between affected family members).
- Kanski's Clinical Ophthalmology, 10th ed., p. 420
- Emery's Elements of Medical Genetics and Genomics
- The Developing Human: Clinically Oriented Embryology, p. 1134
Embryology
The defect results from an arrest of optic cup rim development during approximately the 8th week of gestation. PAX6 acts as a master transcription factor driving differentiation of multiple ocular structures; its dysfunction causes widespread multi-structure dysgenesis rather than isolated iris agenesis.
- The Developing Human: Clinically Oriented Embryology, p. 1134
Clinical Features
Presentation: Typically at birth, with nystagmus and photophobia. Parents often notice apparently large pupils or absence of the colored iris.
Ocular Manifestations (Pan-ocular Disease)
Iris
- Variable severity - from minimal iris hypoplasia (detectable only on retroillumination) to near-total absence
- Gonioscopy typically shows a rudimentary iris frill even in apparently total cases
Cornea (Aniridia-Associated Keratopathy - AAK)
- Tear film instability, dry eye, and epithelial defects
- Limbal stem cell deficiency (LSCD) leads to conjunctivalization of the peripheral cornea (pannus formation)
- End-stage: total central corneal stromal scarring and vascularization
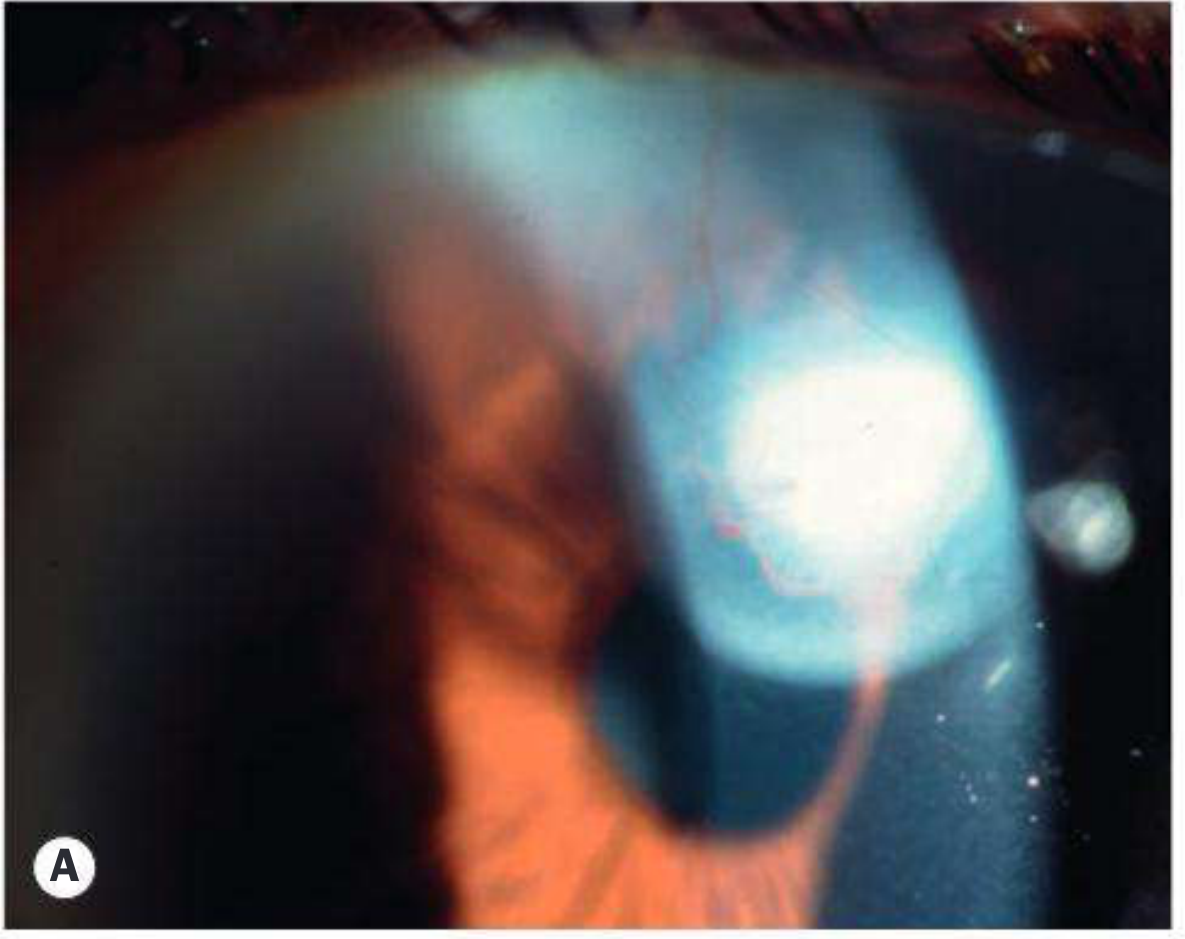
Fig. 11.62A - Advanced aniridia showing corneal vascularization and absent iris (Kanski's)
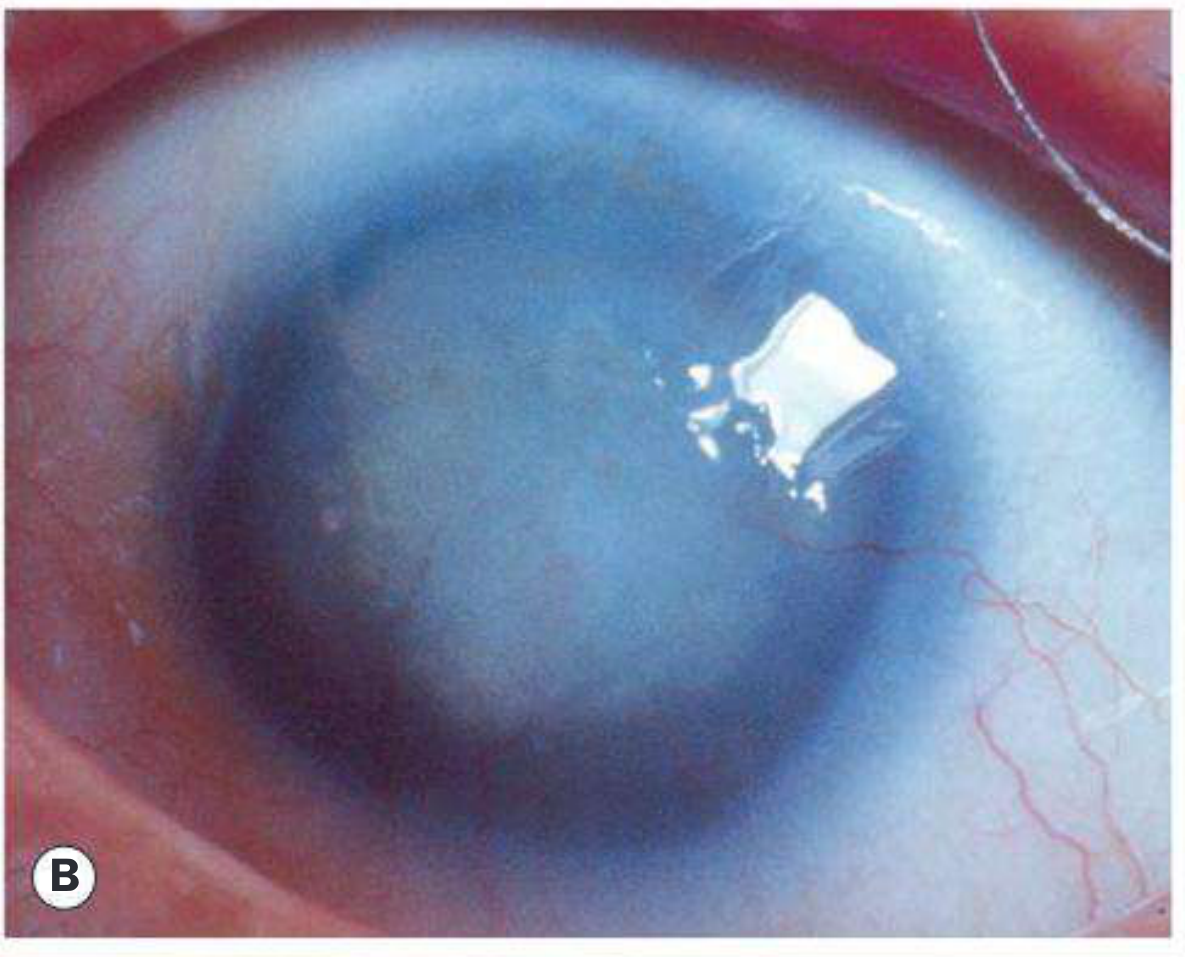
Fig. 11.62B - Near-total aniridia showing dark pupil occupying almost the entire visible eye (Kanski's)
Glaucoma
- Occurs in ~75% of patients
- Usually presents in late childhood or adolescence
- Mechanism: synechial angle closure from contraction of rudimentary iris tissue
- Difficult to manage; prognosis guarded
Foveal and Optic Nerve Hypoplasia
- Foveal hypoplasia is actually a more frequent and reliable clinical sign than visible iris absence (per 2026 European guidelines - Romano et al., Acta Ophthalmol 2026)
- Leads to reduced visual acuity and nystagmus
Lens
- Cataract and lens subluxation may occur
Lids
- Meibomian gland dysfunction is common
WAGR Syndrome
Children with sporadic aniridia have approximately a 30% lifetime risk of Wilms tumor and require systematic surveillance:
- Abdominal ultrasound every 3 months until age 5
- Every 6 months until age 10
- Annually until age 16
- Screening can be stopped if molecular genetic testing confirms absence of WT1 mutation
By contrast, only 1 in 50 Wilms tumor patients has aniridia. The inverse association is important: aniridia is the flag that triggers surveillance; Wilms tumor is not a flag that routinely triggers eye examination.
- Kanski's Clinical Ophthalmology, 10th ed., p. 420
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Management
Glaucoma
- Medical therapy is usually inadequate long-term
- Trabeculectomy with mitomycin C or combined trabeculectomy-trabeculotomy have been tried but usually fail
- Glaucoma drainage devices offer the best chance of long-term IOP control
- Diode laser cycloablation if other modalities fail
Cornea / Ocular Surface
- Frequent lubricants for associated keratopathy
- Limbal stem cell transplantation (with or without keratoplasty) for severe LSCD/corneal failure
Lens
- Cataract surgery often required
- Tinted IOL implant may reduce photophobia
- Limbal trauma must be minimized during any ocular surgery to preserve remaining stem cell function
Vision Rehabilitation
- Painted (tinted/iris-print) contact lenses or simple tinted lenses to create an artificial pupil - can also reduce nystagmus
- Prosthetic iris implantation has been described in pseudophakic eyes, but may worsen glaucoma (use with caution)
Amblyopia / Refractive
- Refractive errors, amblyopia, and strabismus should be managed aggressively
Systemic
-
Gillespie syndrome: cerebellar rehabilitation, learning support
-
WAGR: Wilms tumor surveillance protocol (above); also screen for genitourinary anomalies and monitor intellectual development
-
Kanski's Clinical Ophthalmology, 10th ed., pp. 420-421
Differential Diagnosis
| Condition | Key Distinguishing Feature |
|---|---|
| Axenfeld-Rieger syndrome | Posterior embryotoxon + iris strands; PITX2/FOXC1 mutations; dental/skeletal anomalies |
| Peters anomaly | Central corneal opacity + iris strands to posterior corneal defect |
| Traumatic mydriasis/iridodialysis | History of trauma; unilateral |
| Congenital idriodialysis | Sector defect, not bilateral absence |
Recent Evidence (2026)
The European COST Action ANIRIDIA-NET published the first multinational clinical guidelines in 2026, formalizing that:
- Foveal hypoplasia - not iris absence - is the most frequent and reliable clinical sign
- PAX6 is the main but not the only causative gene
- Management is challenging due to lack of targeted therapies
- Both medical and non-medical approaches (low vision aids, tinted lenses, psychological support) are formalized components of care
Romano et al., Acta Ophthalmol 2026 (PMID 40892309) - European ANIRIDIA-NET Guidelines
Summary Points
- Aniridia = PAX6 mutation causing pan-ocular disorder (iris + fovea + cornea + angle + lens + optic nerve)
- Autosomal dominant (2/3) has no systemic risk; sporadic (1/3) carries ~30% Wilms tumor risk
- WAGR = Wilms tumor + Aniridia + Genital abnormalities + Retardation (chr 11p13 deletion)
- Gillespie syndrome = AR, non-PAX6, + cerebellar ataxia (rare, 1%)
- Glaucoma occurs in 75% - drainage devices are treatment of choice
- Aniridia-associated keratopathy from limbal stem cell deficiency is a major cause of visual loss
- Sporadic cases need renal ultrasound surveillance every 3 months until age 5
This is a shared conversation. Sign in to Orris to start your own chat.