Lymphocyte disorders pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive, textbook-grounded overview of lymphocyte disorders pathology, drawn primarily from Robbins, Cotran & Kumar - Pathologic Basis of Disease and Henry's Clinical Diagnosis and Management by Laboratory Methods.
Lymphocyte Disorders - Pathology
Normal Lymphocyte Morphology (Quick Reference)
Lymphocytes are mononuclear cells, 6-15 µm in diameter, with a single round nucleus showing heavy chromatin blocks staining dark blue on Wright's stain, and scant pale-blue cytoplasm. They average 34% of all leukocytes (normal count: 1.5-4 × 10⁹/L in adults). Large lymphocytes with faint granules represent activated or NK cells. Plasma cells are the terminally differentiated antibody-secreting derivative, with eccentric nuclei, clock-face chromatin, and a prominent perinuclear (Golgi) zone. - Henry's Clinical Diagnosis, p. 643
Classification of Lymphocyte Disorders
Lymphocyte disorders fall into two main groups:
- Reactive/Quantitative disorders - non-neoplastic changes in number or morphology
- Neoplastic (lymphoid) disorders - malignant clonal proliferations
I. Reactive Lymphocyte Disorders
Lymphocytosis
- Absolute lymphocyte count > 4 × 10⁹/L
- Causes: viral infections (EBV, CMV, pertussis, hepatitis), stress responses
- Atypical (reactive) lymphocytes are large, with abundant cytoplasm, indented by surrounding RBCs - characteristic of infectious mononucleosis
Lymphocytopenia
- Count < 1.5 × 10⁹/L
- Causes: corticosteroids, HIV infection, immunosuppressive therapy, autoimmune diseases (SLE), radiation
Hemophagocytic Lymphohistiocytosis (HLH)
A life-threatening systemic condition driven by uncontrolled activation of macrophages and CD8+ cytotoxic T cells.
- Pathogenesis: Familial forms have mutations in CTL/NK cytotoxic granule machinery (perforin, granzymes), impairing killing of infected cells. This triggers persistent T-cell/NK activation with IFN-γ secretion, causing macrophage hyperactivation ("cytokine storm" with IFN-γ, TNF-α, IL-6, IL-12).
- Clinical: Acute febrile illness, splenomegaly/hepatomegaly, cytopenias, hyperferritinemia, elevated soluble IL-2R, high triglycerides, DIC.
- Common trigger: EBV infection; also complicates peripheral T-cell lymphoma.
- Treatment: Glucocorticoids + etoposide; HSC transplantation for familial/refractory cases. Without treatment, median survival < 2 months. - Robbins, p. 553
II. Neoplastic Lymphocyte Disorders (Lymphoid Neoplasms)
General Principles of Pathogenesis
- Most white cell neoplasms carry nonrandom chromosomal translocations or mutations that serve as driver events.
- Oncoproteins block normal maturation (differentiation arrest) or activate pro-survival/growth pathways (RAS/PI3K/AKT).
- Environmental contributors: radiation, chemical carcinogens, viruses (EBV, HTLV-1, H. pylori), immunosuppression.
- Hematologic malignancies are genetically simpler than solid tumors, partly explaining their higher curability. - Robbins, p. 553-554
A. Precursor (Immature) Lymphoid Neoplasms
Acute Lymphoblastic Leukemia/Lymphoma (ALL/LBL)
- Most common cancer in children; ~75-80% are B-cell origin (B-ALL), remainder T-cell.
- Tumor cells are lymphoblasts with arrested differentiation that accumulate as nonfunctional blasts in bone marrow.
- Key genetic lesions:
- t(12;21) - ETV6-RUNX1 fusion (most common in childhood B-ALL, favorable)
- t(9;22) - BCR-ABL (Philadelphia chromosome; 190 kDa protein with stronger tyrosine kinase than the 210 kDa CML form; worse prognosis but responds to TKIs)
- Other cryptic TK rearrangements ("Ph-like ALL") in adults
- Clinical: Bone marrow failure (anemia, thrombocytopenia, neutropenia), rapidly growing masses, CNS involvement
- Treatment: Intensive chemotherapy; CAR-T cells directed against CD19 for relapsed B-ALL; TKI combinations for Ph+ ALL.
- Prognosis is generally excellent in children (>90% cure rate); more guarded in adults. - Robbins, p. 558-559
B. Peripheral (Mature) B-Cell Neoplasms
1. Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma (CLL/SLL)
- Most common adult leukemia in the Western world; ~15,000 new cases/year in the US.
- Median age at diagnosis: 60 years; male predominance (2:1).
- CLL = peripheral blood lymphocytosis > 5000/mm³; SLL = same clone but primarily nodal.
Pathogenesis:
- Chromosomal translocations are rare; dominant anomalies: del(13q14.3), del(11q), del(17p), trisomy 12q.
- Del(13q) targets miR-15a/miR-16-1 (tumor suppressor miRNAs) → BCL2 overexpression → anti-apoptosis.
- NOTCH1 gain-of-function mutations in 10-18% → worse prognosis.
- Unmutated Ig genes (naive B-cell origin) = more aggressive than somatic hypermutated cases.
- Growth depends on BTK signaling via the B-cell receptor → BTK inhibitors (ibrutinib) are highly effective.
Morphology:
- Lymph nodes diffusely effaced by small round lymphocytes (6-12 µm), condensed chromatin, scant cytoplasm.
- Characteristic "proliferation centers" (pale-staining aggregates of larger activated cells) - pathognomonic for CLL/SLL.
- "Smudge cells" in peripheral blood smear (fragile CLL cells disrupted during smear preparation).
- Bone marrow, spleen, and liver almost always involved.
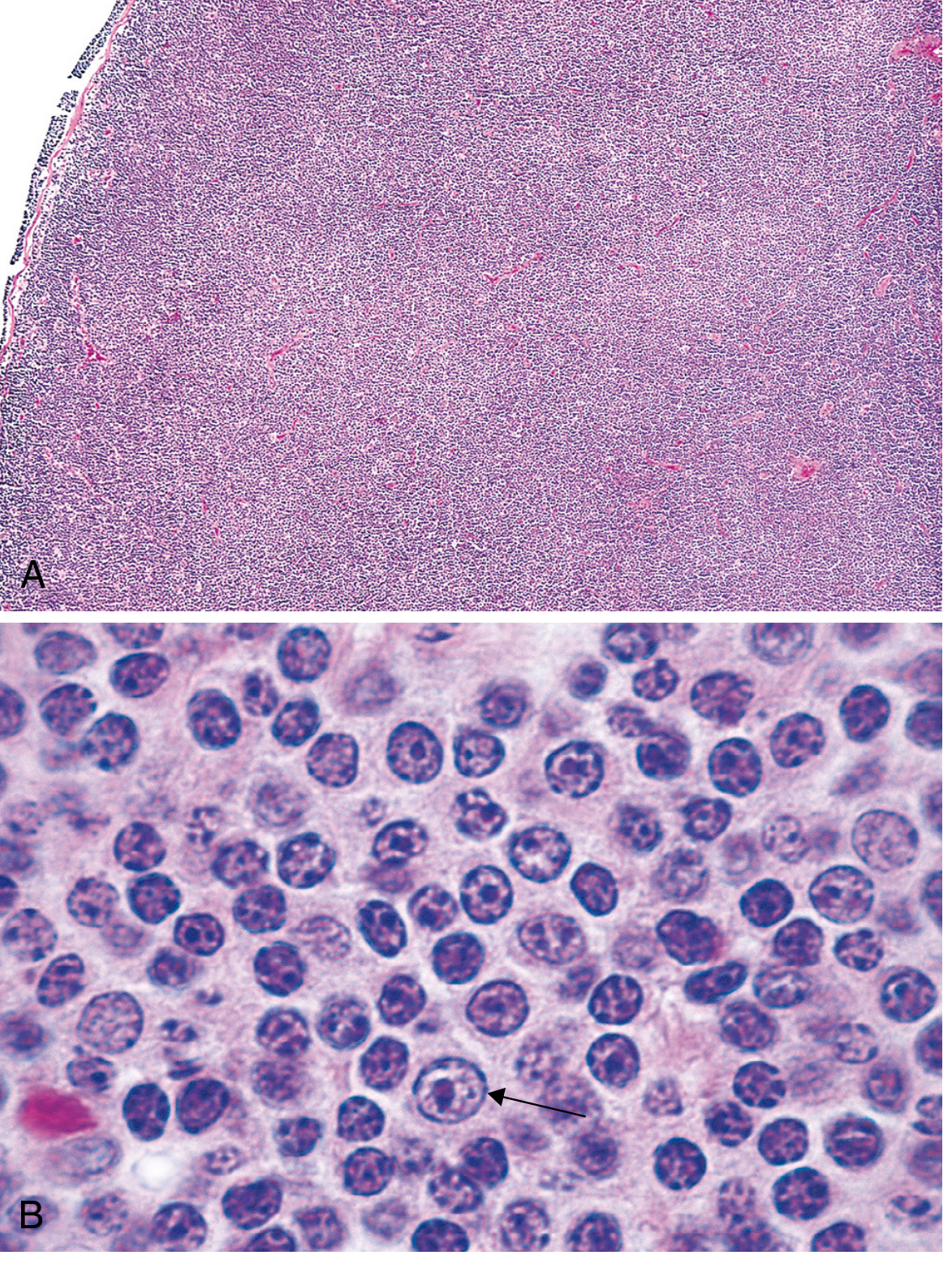
Fig. 13.7 - Small lymphocytic lymphoma/CLL. (A) Low-power: diffuse nodal effacement. (B) High-power: small round lymphocytes with a prolymphocyte (arrow). - Robbins
Immunophenotype: CD5+, CD23+, CD19+, CD20 (dim), IgM/IgD (dim) - the CD5 positivity is a key diagnostic feature (normally a T-cell marker).
Complications:
- Hypogammaglobulinemia → recurrent infections (most common cause of death)
- Autoimmune hemolytic anemia
- Transformation to diffuse large B-cell lymphoma (Richter syndrome, ~5% of cases)
2. Follicular Lymphoma
- Second most common NHL in adults (US); arises from germinal center B cells.
- Hallmark: t(14;18) translocation → BCL2 overexpression (BCL2 normally suppresses apoptosis; inappropriately expressed in follicular lymphoma, conferring survival advantage)
- Morphology: Nodular (follicular) growth pattern; cells resemble centrocytes (small, cleaved) and centroblasts (large, noncleaved).
- Immunophenotype: CD10+, BCL6+, BCL2+, CD20+, CD19+ (unlike normal reactive germinal center B cells, which are BCL2 negative)
- Typically indolent but incurable with standard therapy; may transform to DLBCL (~30-40% over time). - Robbins, p. 562
3. Diffuse Large B-Cell Lymphoma (DLBCL)
- Most common NHL (~25-30% of all NHLs in adults).
- Clinically aggressive; arises de novo or via transformation from indolent lymphomas.
- Two major molecular subtypes (gene expression profiling):
- Germinal center B-cell (GCB) type - better prognosis
- Activated B-cell (ABC) type - worse prognosis; depends on constitutive NF-κB activation
- Morphology: Diffuse sheets of large lymphoid cells with vesicular nuclei and prominent nucleoli.
- Treatment: R-CHOP (rituximab + cyclophosphamide, doxorubicin, vincristine, prednisone); ~60-70% cure rate.
4. Burkitt Lymphoma
- Highly aggressive; associated with t(8;14) translocation → MYC overactivation.
- Three forms: endemic (Africa, EBV-driven, jaw/facial bones), sporadic (abdomen), immunodeficiency-associated (HIV).
- Morphology: Medium-sized cells with basophilic cytoplasm; classic "starry-sky" pattern (macrophages engulfing apoptotic tumor cells give light areas amid dark tumor cells).
- Fastest-dividing human tumor (near 100% Ki-67 proliferation index).
- Curable with intensive chemotherapy if caught early.
5. Marginal Zone Lymphomas / MALToma
- Arise within lymph nodes, spleen, or extranodal tissues (mucosa-associated lymphoid tissue).
- Extranodal MALToma is special:
- Arises at sites of chronic inflammation (autoimmune or infectious): stomach (H. pylori), salivary gland (Sjögren syndrome), thyroid (Hashimoto thyroiditis)
- Remains localized for prolonged periods
- May regress with eradication of the inciting agent (e.g., antibiotic treatment of H. pylori can lead to gastric MALToma regression)
- Progression involves chromosomal translocations activating NF-κB via BCL10/MALT1 upregulation: t(11;18), t(14;18), t(1;14)
- Cells of origin: memory B cells (somatically hypermutated Ig genes)
6. Hairy Cell Leukemia
- Rare B-cell neoplasm; ~2% of leukemias; predominantly middle-aged White males (median age 55; M:F = 5:1).
- Pathogenesis: >90% harbor BRAF V600E activating mutation (same mutation as in melanoma and Langerhans cell histiocytosis).
- Morphology: Tumor cells have fine hair-like cytoplasmic projections (best seen on phase-contrast microscopy or electron microscopy); tartrate-resistant acid phosphatase (TRAP) stain positive.
- Presents with massive splenomegaly, pancytopenia, and "dry tap" on bone marrow aspiration (fibrosis).
- Highly responsive to purine analogs (cladribine, pentostatin); BRAF inhibitors (vemurafenib) for refractory cases.
C. Plasma Cell Neoplasms (Terminal B-Cell Differentiation)
Multiple Myeloma
- Clonal expansion of plasma cells in bone marrow, producing monoclonal immunoglobulin (M protein).
- Pathogenesis: Primary translocations involving Ig heavy-chain locus (14q32) with partners including FGFR3 (t(4;14)), Cyclin D1 (t(11;14)), MMSET.
- Clinical features (mnemonic CRAB): hyperCalcemia, Renal failure, Anemia, Bone lesions (punched-out lytic lesions).
- Secondary infections due to hypogammaglobulinemia of normal Ig isotypes.
- Bence-Jones proteins (free light chains in urine) cause cast nephropathy.
- Bone marrow: >10% plasma cells; abnormal plasma cells with "clock-face" chromatin.
D. Mature T-Cell and NK-Cell Neoplasms
Peripheral T-Cell Lymphoma (PTCL), Unspecified
- Most common mature T-cell lymphoma; aggressive; median age ~60 years.
- Diffuse nodal infiltrate of atypical T cells with variable size; CD3+, CD4+ or CD8+.
- Associated with systemic symptoms, eosinophilia, HLH.
- Poor prognosis; responds poorly to R-CHOP.
Adult T-Cell Leukemia/Lymphoma (ATLL)
- Caused by HTLV-1 retrovirus (endemic in Japan, Caribbean, Central Africa).
- Highly aggressive; skin lesions, hypercalcemia, lytic bone lesions.
- Hallmark cell: "flower cell" (multilobated nucleus) in peripheral blood.
Mycosis Fungoides / Sézary Syndrome
- Primary cutaneous T-cell lymphoma; presents as chronic skin rash progressing through patch → plaque → tumor stages.
- Sézary syndrome = leukemic form (Sézary cells = large lymphocytes with cerebriform/convoluted nuclei in blood).
- Neoplastic cells are CD4+ skin-homing T cells.
Anaplastic Large Cell Lymphoma (ALCL)
- Large anaplastic T cells (or null phenotype); characteristic "hallmark cells" - horseshoe or kidney-shaped nuclei.
- ALK-positive ALCL: harbors t(2;5) producing NPM-ALK fusion protein; primarily young patients; excellent prognosis.
- ALK-negative ALCL: older patients; worse prognosis.
- Strongly CD30+. Treatment includes brentuximab vedotin (anti-CD30 antibody-drug conjugate).
E. Hodgkin Lymphoma (HL)
A distinctive lymphoma defined by Reed-Sternberg (RS) cells (large binucleate cells with prominent "owl-eye" nucleoli) in a background reactive infiltrate. RS cells are clonally derived from germinal center B cells but have lost B-cell identity (absent Ig expression). CD15+, CD30+.
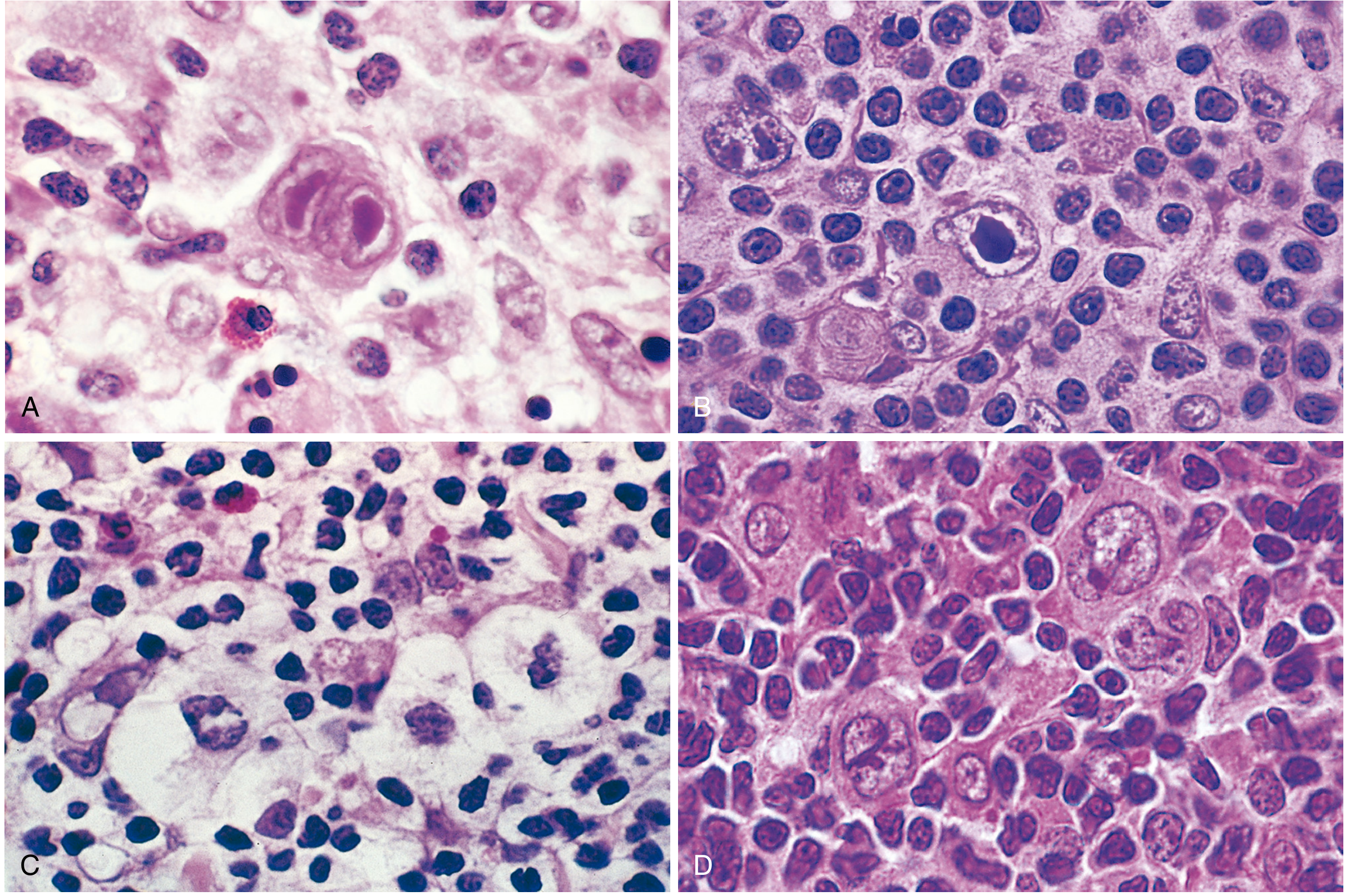
Fig. 13.25 Reed-Sternberg cells and variants. (A) Diagnostic RS cell with two nuclear lobes and "owl-eye" nucleoli. (B) Mononuclear variant. (C) Lacunar variant in open retracted space. (D) Lymphohistiocytic (L&H/"popcorn") variant. - Robbins
WHO Classification of Hodgkin Lymphoma
| Subtype | Frequency | Key Morphology | EBV | Immunophenotype | Clinical |
|---|---|---|---|---|---|
| Nodular Sclerosis | 65-70% (most common) | Collagen bands dividing nodules; lacunar RS cells | Usually negative | CD15+, CD30+ | Young adults; mediastinum; equal sex; Stage I-II |
| Mixed Cellularity | 20-25% | Diffuse heterogeneous infiltrate; abundant RS cells; eosinophils, plasma cells | 70% | CD15+, CD30+ | Males; Stage III-IV; systemic symptoms; biphasic incidence |
| Lymphocyte-Rich | Uncommon | Reactive lymphocytes predominate; few RS cells | 40% | CD15+, CD30+ | Older males; good prognosis |
| Lymphocyte-Depleted | <5% (rarest) | Paucity of lymphocytes; abundant pleomorphic RS cells | >90% | CD15+, CD30+ | Older males, HIV+, developing countries; advanced disease; worst prognosis |
| Nodular Lymphocyte Predominant (NLPHL) | ~5% | "Popcorn" L&H cells in B-cell follicle background; no classic RS cells | Rarely | CD20+, BCL6+; CD15−, CD30− | Young males; cervical/axillary; excellent prognosis; can transform to DLBCL |
Staging and prognosis: Spread is contiguous (node → spleen → liver → marrow). Stages I/IIA: ~90% cure rate. Stages IVA/IVB: 60-70% 5-year disease-free survival. Long-term survivors from radiotherapy have increased risk of secondary malignancies (lung cancer, melanoma, breast cancer). - Robbins, p. 571-576
Summary Table - Key Distinguishing Features
| Disorder | Cell Origin | Key Molecular Lesion | Hallmark Feature |
|---|---|---|---|
| B-ALL | Precursor B cell | BCR-ABL (t9;22), ETV6-RUNX1 (t12;21) | Blasts in marrow; differentiation arrest |
| CLL/SLL | Mature naive/memory B cell | del(13q), BCL2 overexpression | Smudge cells; CD5+CD23+ B cells; proliferation centers |
| Follicular lymphoma | Germinal center B cell | t(14;18), BCL2 | Follicular growth; BCL2+ germinal centers |
| DLBCL | Germinal center/activated B cell | MYC, BCL2, BCL6 | Diffuse large cells; aggressive |
| Burkitt lymphoma | Germinal center B cell | t(8;14), MYC | Starry-sky pattern; fastest growing tumor |
| MALToma | Memory B cell | BCL10/MALT1 (NF-κB) | H. pylori/chronic inflammation driven; regresses with antigen elimination |
| Hairy cell leukemia | Memory B cell | BRAF V600E | Hair-like projections; TRAP+; splenomegaly |
| Multiple myeloma | Plasma cell | t(4;14), t(11;14) | M-spike; CRAB features |
| ALCL | T cell / null | NPM-ALK t(2;5) | Hallmark cells; CD30+; ALK+ = better prognosis |
| Hodgkin lymphoma | Germinal center B cell | Unknown (epigenetic) | Reed-Sternberg cells; CD15+CD30+ |
| HLH | CD8+ T cell / NK cell | Perforin/granzyme mutations (familial) | Cytokine storm; hemophagocytosis |
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (9780443264528); Henry's Clinical Diagnosis and Management by Laboratory Methods (9780323673204)
This is a shared conversation. Sign in to Orris to start your own chat.