Take required standard resources and give brief explanation of pharmacokinetics of iv agents in view of md anesthesia
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Loading Image
Reading File
I now have excellent content from authoritative sources. Let me compile the comprehensive answer:
Pharmacokinetics of Intravenous (IV) Anesthetic Agents - MD Anesthesia
Sources: Barash, Cullen & Stoelting's Clinical Anesthesia 9e; Miller's Anesthesia 10e; Morgan & Mikhail's Clinical Anesthesiology 7e
1. Why Standard PK Concepts Differ for IV Anesthetics
In most clinical pharmacology, dosing occurs over days-to-weeks, so simple one-compartment models (total Vd, elimination half-life) suffice. In the OR, however, onset, maintenance, and offset are measured in minutes - making distribution kinetics, not just elimination, the dominant driver of drug behavior. IV anesthetics are highly lipophilic, extensively distribute across tissue compartments, and have narrow two-tailed therapeutic indices (underdose = awareness; overdose = hemodynamic collapse). This demands multicompartment modeling.
(Barash, Clinical Anesthesia, p.1465; Miller's Anesthesia, p.789)
2. The Three-Compartment Model
The standard pharmacokinetic framework for IV anesthetics uses a three-compartment model:
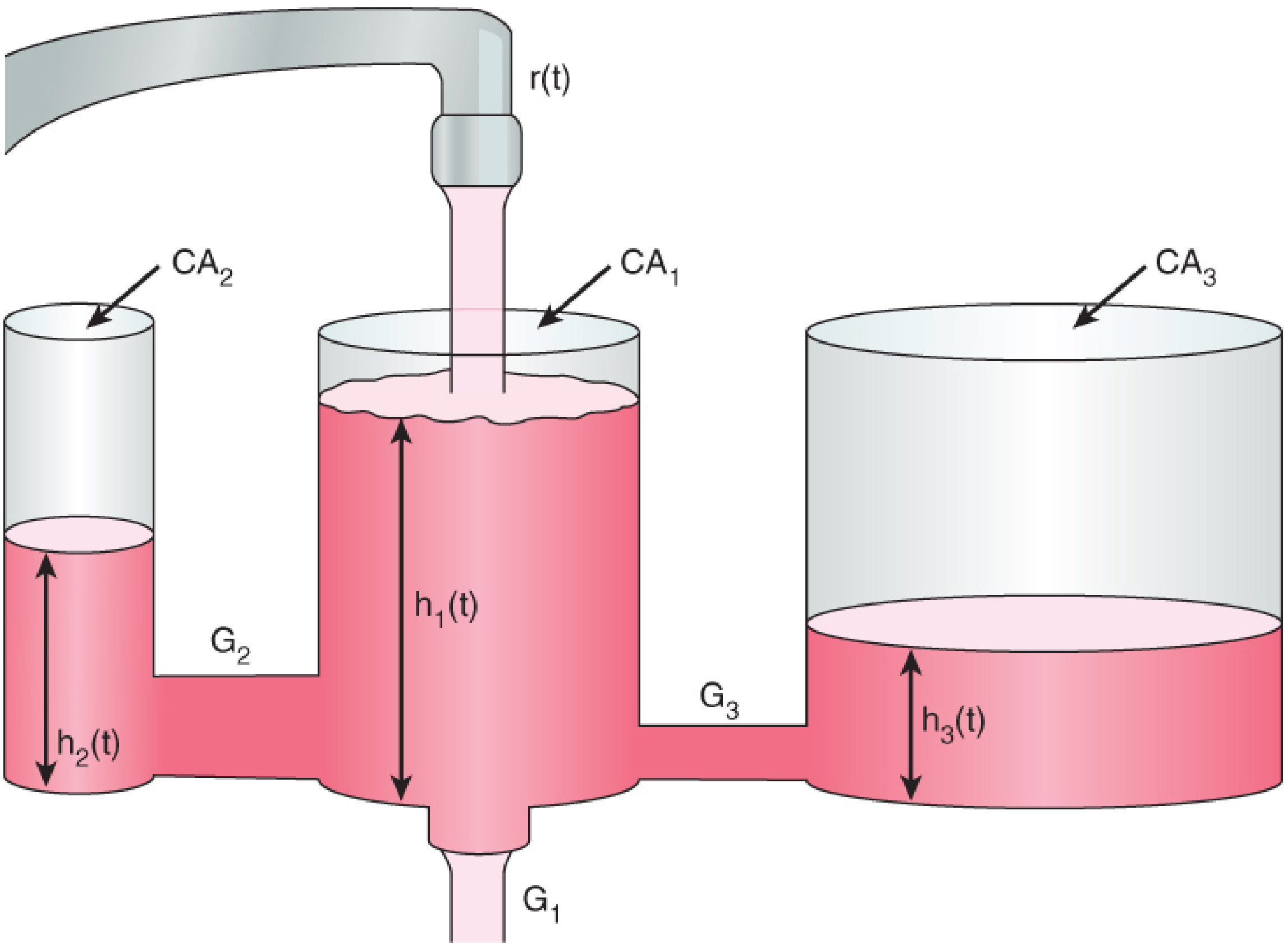
Hydraulic analogy of the three-compartment model - Barash Clinical Anesthesia 9e
| Compartment | What it represents | Rate of exchange |
|---|---|---|
| Central (V1) | Blood/plasma, highly perfused organs (brain, heart, liver) | Drug arrives here first |
| Fast peripheral (V2) | Muscle, gut - moderate perfusion | Rapid exchange (minutes) |
| Slow peripheral (V3) | Fat, bone - poorly perfused | Slow exchange (hours) |
After a bolus: plasma concentration falls in three phases:
- Alpha (α) phase - rapid distribution from blood to peripheral compartments (minutes)
- Beta (β) phase - slower redistribution and continued distribution (tens of minutes)
- Gamma (γ) phase - true elimination (hours)
This triexponential decay is described mathematically as:
C(t) = A·e^(-αt) + B·e^(-βt) + C·e^(-γt)
Key insight: The brief duration of action of a single bolus of propofol or thiopental is NOT due to metabolism - it is due to redistribution from brain to peripheral compartments. Metabolism becomes relevant only during prolonged infusion.
(Barash, p.1468-1469)
3. Context-Sensitive Half-Time (CSHT) - The Most Clinically Relevant PK Concept
Definition: The time required for plasma concentration to decrease by 50% after stopping a continuous infusion - where "context" refers to the duration of infusion.
For a one-compartment drug, this equals the elimination half-life regardless of infusion duration. For multicompartment drugs (all IV anesthetics), CSHT increases with infusion duration as peripheral compartments become saturated and begin returning drug to plasma.
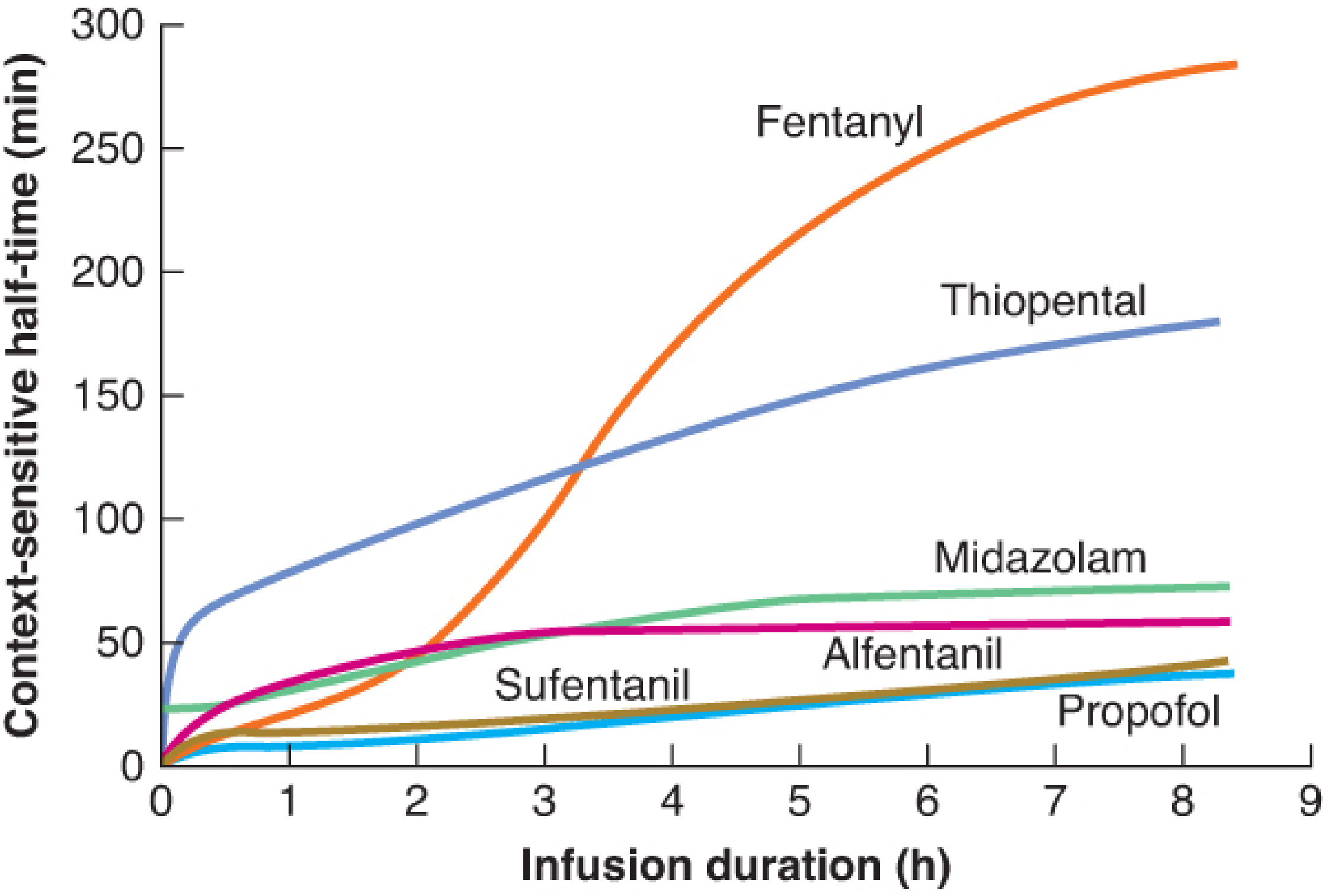
Reading this graph:
- Propofol - CSHT remains <40 min even after 8 hours of infusion → predictable recovery regardless of infusion duration → ideal for TIVA
- Fentanyl - CSHT rises steeply after ~2 hours → prolonged infusion leads to unpredictable, extended recovery
- Thiopental - large early CSHT (V3 accumulation in fat) → unsuitable as an infusion
- Remifentanil (not shown) - CSHT remains ~3-5 min at any duration due to ester hydrolysis in plasma/tissues - the true "context-insensitive" opioid
(Barash, p.1469; Miller's, p.3738-3746)
4. Key PK Parameters Relevant to IV Anesthetics
| Parameter | Definition | Clinical relevance |
|---|---|---|
| Volume of distribution (Vd) | Apparent volume drug occupies if uniformly distributed at plasma concentration | Highly lipophilic drugs (propofol, fentanyl) have large Vd - large loading doses needed |
| Clearance (Cl) | Volume of plasma cleared of drug per unit time | Determines maintenance infusion rate at steady state |
| t½ elimination | 0.693 × Vd/Cl | Useful for single bolus; misleading for infusions |
| Effect-site (Ce) | Concentration at target organ (brain) | Lags behind plasma concentration - explains hysteresis |
| ke0 | Rate constant for plasma-to-effect-site equilibration | Higher ke0 = faster onset; propofol ke0 >> ketamine ke0 |
| t½ke0 | Time for effect-site to equilibrate with plasma | Propofol ~2.6 min; fentanyl ~4 min; alfentanil ~1 min |
5. Pharmacokinetics of Individual IV Agents
Propofol (2,6-diisopropylphenol) - the "gold standard"
- Distribution: Initial t½ 1-8 min (rapid), secondary t½ 30-70 min, elimination t½ 2-24 h
- Clearance: 20-30 mL/kg/min - exceeds hepatic blood flow (15 mL/kg/min), implying extrahepatic metabolism (kidneys, lungs contribute ~30%)
- CSHT: <40 min at any infusion duration up to 8 hours
- Protein binding: >97% (albumin, α1-acid glycoprotein)
- Vd (steady state): ~4 L/kg
- Clinical implication: Low CSHT + rapid hepatic + extrahepatic clearance makes it ideal for TIVA, ICU sedation, and outpatient anesthesia. Liver/renal disease do not significantly alter kinetics.
(Barash, p.1474-1475)
Thiopental (barbiturate) - historical comparator
- Onset: Rapid (one arm-brain circulation, ~20 sec) due to high lipophilicity
- Duration of bolus: Short (5-8 min) due to redistribution - NOT metabolism
- CSHT: Very high - rises steeply with infusion duration (large V3 fat compartment); unsuitable for infusion
- Elimination t½: 10-12 hours
- Why it fell out of favour: Long CSHT, no antiemetic properties, porphyria precaution
Etomidate
- Distribution: Rapid onset (one arm-brain)
- Protein binding: ~75%
- Metabolism: Rapid hepatic ester hydrolysis → inactive metabolites; also plasma cholinesterase
- t½ elimination: ~3 hours
- Key PK feature: Cardiovascular stability due to minimal effect on SVR
- Limitation: Adrenocortical suppression (inhibits 11β-hydroxylase) - single dose suppresses cortisol for 6-12 hours
Ketamine
- Highly lipophilic → rapid CNS uptake
- Vd: ~3 L/kg
- Duration of bolus: 10-15 min (redistribution)
- Metabolism: Hepatic (CYP3A4, CYP2B6) → norketamine (still ~1/3 potency) → further hydroxylation
- t½: ~2-3 hours
- Key PK feature: Norketamine (active metabolite) prolongs effects with repeated dosing; norketamine may explain prolonged analgesia
- Clinical use: Low dose infusion (0.1-0.5 mg/kg/h) for opioid-sparing analgesia - norketamine accumulation is a PK consideration
Midazolam
- Highly protein bound (~94%)
- Hepatic metabolism: CYP3A4 → 1-hydroxymidazolam (active) then glucuronidation
- t½: 1.5-3.5 hours
- CSHT: Moderate but rising (~75 min after 8h infusion) - slower offset than propofol
- Dose adjustment: Reduce in hepatic impairment, elderly (reduced CYP3A4), and hypoalbuminaemia
Dexmedetomidine
- Protein binding: ~94%
- Hepatic metabolism: Glucuronidation + CYP2A6 oxidation
- t½: ~2 hours
- Vd: ~118 L
- CSHT: Context-sensitive accumulation relevant with prolonged ICU infusions
- Key feature: Does not cause respiratory depression at sedative doses
6. Effect-Site (Biophase) Pharmacokinetics
Plasma concentration does not equal effect-site concentration. There is always a hysteresis (lag) between peak plasma levels and peak clinical effect. This is quantified by ke0 (the equilibration rate constant between plasma and effect site) and its derived t½ke0.
Clinical example: After a propofol bolus:
- Peak plasma: ~1-2 minutes
- Peak effect (EEG change, BIS drop): ~3-4 minutes
This means dosing should be guided by effect-site kinetics, not plasma kinetics. Target-controlled infusion (TCI) pumps use validated PK models (e.g., Marsh or Schnider for propofol; Minto for remifentanil) to calculate real-time infusion rates targeting either plasma (Cp) or effect-site (Ce) concentration.
7. Target-Controlled Infusion (TCI) and PK Modeling
TCI systems use the three-compartment model parameters to continuously calculate infusion rates needed to achieve and maintain a user-specified target concentration. Key concepts:
- Bolus-elimination-transfer (BET) scheme: An initial bolus fills V1 to target; a higher-rate infusion replaces eliminated drug; rate progressively reduces as V2/V3 saturate
- Schnider model (propofol): Uses patient covariates (age, weight, height, lean body mass) to individualize PK parameters
- Minto model (remifentanil): Age and lean body mass as covariates
- TIVA advantage: Predictable depth with propofol + remifentanil CSHT - both agents have low/flat CSHT curves, ensuring reliable offset
(Barash, p.1473; Miller's p.3738-3746)
8. Special Populations - PK Alterations
| Population | Key change | Practical adjustment |
|---|---|---|
| Elderly | Reduced Vd, reduced hepatic clearance, lower albumin | Reduce induction dose by 30-50%; slower infusion rates |
| Obesity | Increased V3 (fat), altered protein binding | Loading dose based on LBM; propofol clearance relatively preserved |
| Hepatic failure | Reduced Phase I metabolism (CYP), reduced albumin | Increased free fraction of protein-bound drugs; reduce dose; avoid etomidate continuous infusion |
| Renal failure | Reduced excretion of active/polar metabolites | Norketamine and 1-OH midazolam can accumulate |
| Paediatric | Higher Vd, faster clearance per kg | Higher mg/kg induction dose; faster infusion rates for propofol |
9. Rise to Steady-State Concentration
During a constant infusion, concentration rises toward Css (steady state) following:
Cp(t) = Css [1 - e^(-kt)]
- Reaches 50% Css in 1 elimination half-life
- Reaches 90% Css in ~3.3 half-lives
- For multicompartment drugs, true steady state takes much longer (V2 and V3 must fill)
This is why a simple maintenance infusion without an initial bolus takes too long to reach therapeutic effect - a bolus to fill V1, then adjusted infusion rates are required.
(Miller's Anesthesia, p.789-790)
Summary Table: PK Properties of Key IV Agents
| Drug | Vd (L/kg) | Cl (mL/kg/min) | t½ elim | CSHT (8h) | Protein binding | Metabolism |
|---|---|---|---|---|---|---|
| Propofol | ~4 | 20-30 | 2-24 h | ~40 min | >97% | Hepatic + extrahepatic |
| Thiopental | ~2.5 | 3-4 | 10-12 h | ~180 min | ~85% | Hepatic (CYP2C19) |
| Etomidate | ~4.5 | 18-25 | ~3 h | Moderate | ~75% | Hepatic ester hydrolysis |
| Ketamine | ~3 | 12-17 | ~2-3 h | Moderate | ~12-47% | Hepatic (CYP3A4) |
| Midazolam | ~1.1 | 6-8 | 1.5-3.5 h | ~75 min | ~94% | Hepatic (CYP3A4) |
| Remifentanil | ~0.35 | 40-60 | ~10 min | ~3-5 min | ~70% | Plasma esterases |
| Dexmedetomidine | ~118 L | ~39 | ~2 h | Accumulates | ~94% | Hepatic glucuronidation |
Key MD Anesthesia Points to Remember:
- Redistribution (not metabolism) explains short action of single-bolus lipophilic drugs
- CSHT is the correct metric for infusion pharmacokinetics - not elimination half-life
- Effect-site lag (ke0/t½ke0) must be accounted for to avoid overdose during induction
- Propofol's extrahepatic metabolism explains why its kinetics remain predictable in liver/renal disease
- TCI pumps operationalize three-compartment models in real time - understanding the underlying PK is essential for troubleshooting and manual dosing
- Remifentanil is the only IV anesthetic/analgesic with truly context-insensitive kinetics (plasma ester hydrolysis, CSHT ~3-5 min regardless of duration)
(Barash, Cullen & Stoelting's Clinical Anesthesia 9e, Ch.19; Miller's Anesthesia 10e, Ch.11 & 23; Morgan & Mikhail's Clinical Anesthesiology 7e, Ch.11)
This is a shared conversation. Sign in to Orris to start your own chat.