Pathology of Acute kidney injury
Reading File
search_medical_text
acute kidney injury pathology etiology classification mechanisms
search_medical_text
acute tubular necrosis histopathology ischemic nephrotoxic AKI
search_medical_image
acute kidney injury pathology histology tubular necrosis
GradeResponse
GradeResponse
GradeResponse
search_medical_text
prerenal intrinsic postrenal AKI pathophysiology GFR tubular function
search_medical_text
AKI biomarkers diagnosis KDIGO creatinine oliguria stages
GradeResponse
GradeResponse
Here is a comprehensive overview of the pathology of Acute Kidney Injury (AKI):
Pathology of Acute Kidney Injury (AKI)
Definition
AKI is a clinical syndrome characterized by an abrupt decline in glomerular filtration rate (GFR) with consequent retention of nitrogenous waste products (urea, creatinine) and disruption of fluid, electrolyte, and acid-base homeostasis.
KDIGO Diagnostic Criteria — any of the following:
- Rise in serum creatinine ≥0.3 mg/dL within 48 hours
- Rise in serum creatinine ≥1.5× baseline within 7 days
- Urine output <0.5 mL/kg/hr for >6 consecutive hours
Classification & KDIGO Staging
| Stage | Serum Creatinine | Urine Output |
|---|---|---|
| 1 | 1.5–1.9× baseline OR ≥0.3 mg/dL rise | <0.5 mL/kg/hr for 6–12 h |
| 2 | 2.0–2.9× baseline | <0.5 mL/kg/hr for ≥12 h |
| 3 | ≥3× baseline OR ≥4 mg/dL OR RRT initiated | <0.3 mL/kg/hr for ≥24 h OR anuria for ≥12 h |
Etiologic Classification
The causes of AKI are traditionally divided into three broad categories (Harrison's, p. 8436):
1. Prerenal AKI (~55% of cases)
Pathophysiology: Reduced renal perfusion pressure → activation of renin-angiotensin-aldosterone system (RAAS) and ADH → sodium and water retention → BUN:Cr ratio >20:1. The kidney parenchyma is structurally intact.
Key causes:
- Hypovolemia: hemorrhage, GI losses, burns, diuretic overuse
- Reduced cardiac output: heart failure, cardiogenic shock, massive PE
- Systemic vasodilation: sepsis, cirrhosis (hepatorenal syndrome), anaphylaxis
- Afferent arteriolar constriction: NSAIDs (inhibit prostaglandin-mediated vasodilation), contrast nephropathy, hypercalcemia
- Efferent arteriolar dilation: ACE inhibitors, ARBs (reduce GFR by dropping glomerular efferent pressure — especially dangerous in bilateral renal artery stenosis)
If uncorrected, prerenal azotemia progresses to ischemic intrinsic AKI (ATN).
2. Intrinsic (Renal Parenchymal) AKI (~40%)
The most common causes are sepsis, ischemia, and nephrotoxins (Harrison's, p. 8440). Classified anatomically by the compartment affected:
A. Tubular Disease — Acute Tubular Necrosis (ATN) (most common cause of intrinsic AKI)
Ischemic ATN:
- Sustained hypoperfusion → proximal tubular cells (S3 segment, pars recta) and medullary thick ascending limb most vulnerable due to high metabolic demand and relatively low oxygen delivery
- Cellular mechanisms:
- ATP depletion → failure of Na⁺/K⁺-ATPase → intracellular Na⁺ and Ca²⁺ overload
- Cytoskeletal disruption → loss of brush border and tight junctions → backleak of filtrate
- Apoptosis and necrosis of tubular epithelial cells
- Sloughed cells + debris → intratubular casts → tubular obstruction → ↑ intratubular pressure → ↓ GFR
Nephrotoxic ATN:
- Direct tubular toxicity (aminoglycosides, cisplatin, amphotericin B, heavy metals)
- Pigment nephropathy: hemoglobin (hemolysis) or myoglobin (rhabdomyolysis) → tubular toxicity + obstruction
- Crystal nephropathy: uric acid (tumor lysis syndrome), calcium oxalate, acyclovir, methotrexate
Histopathology of ATN:
- Tubular epithelial cell swelling, vacuolation, loss of brush border
- Intratubular casts (granular, pigmented)
- Patchy tubular cell necrosis with intact basement membrane (allowing regeneration)
- Interstitial edema
- Minimal or absent glomerular and vascular changes
The image below illustrates macroscopic and histological changes in AKI (rhabdomyolysis/nephrotoxic model), showing tubular dilatation, vacuolation, atrophy, and intratubular myoglobin casts:
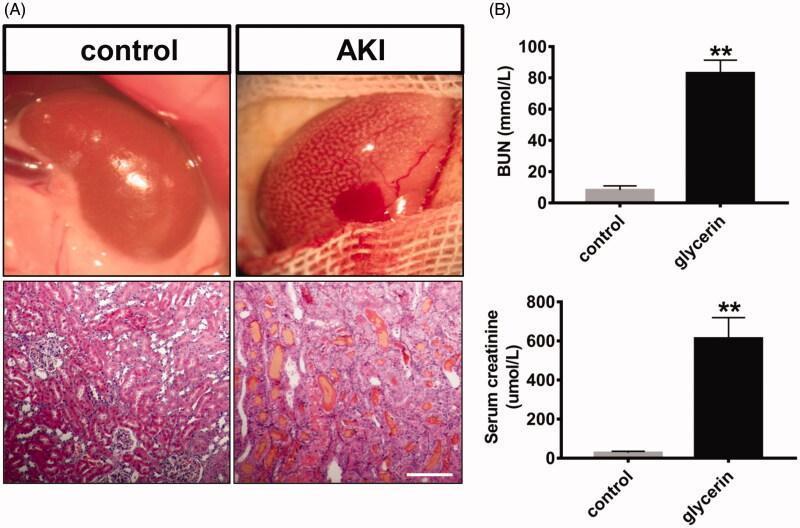
Macroscopic: AKI kidney is enlarged with pale, irregular surface and focal hemorrhage. Histology (H&E): tubular dilatation, vacuolation, cast formation, versus organized tubular architecture in control. Biochemically: marked elevation of BUN (>80 mmol/L) and creatinine (~600 μmol/L).
B. Glomerular Disease
- Rapidly progressive (crescentic) GN: anti-GBM disease (Goodpasture's), ANCA-associated vasculitis, immune complex GN (lupus nephritis, IgA nephropathy with crescents)
- Thrombotic microangiopathy (TMA): TTP-HUS — fibrin thrombi occlude glomerular capillaries → microangiopathic hemolytic anemia + thrombocytopenia + AKI
C. Interstitial Disease — Acute Interstitial Nephritis (AIN)
- Drug-induced (most common): beta-lactam antibiotics, NSAIDs, PPIs, rifampicin, allopurinol
- Infectious: leptospirosis, hantavirus, CMV, EBV
- Autoimmune: sarcoidosis, SLE
- Histopathology: diffuse or patchy lymphocytic and eosinophilic infiltrate in the interstitium, tubulitis (lymphocytes invading tubular epithelium), interstitial edema; glomeruli typically spared
D. Vascular Disease
- Large vessel: bilateral renal artery thrombosis/embolism, aortic dissection, renal vein thrombosis
- Small vessel: polyarteritis nodosa, scleroderma renal crisis, atheroembolic disease (cholesterol crystal emboli — livedo reticularis, eosinophilia, low complement)
3. Postrenal (Obstructive) AKI (~5%)
Mechanism: Urinary tract obstruction → ↑ intratubular back-pressure → ↓ GFR → if bilateral (or unilateral with single functioning kidney)
| Level of Obstruction | Common Causes |
|---|---|
| Ureteric (bilateral) | Retroperitoneal fibrosis, malignancy, bilateral stones, ligation |
| Bladder neck | BPH, prostate/bladder cancer, neurogenic bladder |
| Urethral | Stricture, phimosis |
- Pathophysiology: Early: tubuloglomerular feedback via prostaglandins and angiotensin maintains some GFR. After 12–24 h: renal vasoconstriction, tubular atrophy begins. After weeks: irreversible tubulointerstitial fibrosis.
- Relief of obstruction may be followed by post-obstructive diuresis — massive natriuresis from osmotic solute load and tubular dysfunction.
Biomarkers
Traditional markers — functional but not injury-specific (Harrison's, p. 8462):
- BUN and serum creatinine reflect reduced GFR, not cellular damage; slow to rise
- BUN:Cr ratio >20:1 suggests prerenal; ratio ~10:1 suggests intrinsic
Novel injury biomarkers:
| Biomarker | Compartment | Significance |
|---|---|---|
| NGAL (Neutrophil Gelatinase-Associated Lipocalin) | Urine/plasma | Proximal tubule injury; rises within 2–4 h |
| KIM-1 (Kidney Injury Molecule-1) | Urine | Proximal tubule; ischemic/nephrotoxic ATN |
| IL-18 | Urine | Ischemic ATN |
| Cystatin C | Serum | Earlier GFR marker than creatinine |
| TIMP-2 × IGFBP7 | Urine | Cell cycle arrest; predicts AKI progression |
Urine microscopy: Granular ("muddy brown") casts = ATN. RBC casts = glomerulonephritis. WBC casts = AIN/pyelonephritis. Eosinophiluria = AIN (low sensitivity).
Furosemide stress test: Urine output <200 mL over 2 h after IV furosemide 1.0–1.5 mg/kg predicts progression to severe AKI and need for renal replacement therapy (Harrison's, p. 8462).
Phases of AKI (ATN)
| Phase | Duration | Features |
|---|---|---|
| Initiation | Hours–days | Insult occurs; GFR falls; creatinine begins to rise |
| Extension | Hours–days | Continued ischemia/inflammation; maximal cellular injury |
| Maintenance | 1–2 weeks | GFR nadir; oliguria/anuria; uremic complications |
| Recovery | Days–weeks | Tubular regeneration from viable cells; polyuria (tubular concentrating defect); risk of hyponatremia |
Complications of AKI
- Fluid overload: pulmonary edema, hypertension
- Hyperkalemia: most life-threatening; cardiac arrhythmia
- Metabolic acidosis: anion-gap type (↓ acid excretion)
- Uremia: pericarditis, encephalopathy, platelet dysfunction
- Hyponatremia / Hyperphosphatemia / Hypocalcemia
- Susceptibility to infection: leading cause of death in ICU AKI
Outcomes
- Recovery: Most ATN resolves with tubular regeneration (basement membrane intact); complete recovery in weeks
- CKD progression: ~10–20% of survivors develop de novo or worsened CKD; repeated AKI episodes accelerate CKD
- Mortality: ICU-associated AKI requiring dialysis carries ~50–60% in-hospital mortality
Sources: Harrison's Principles of Internal Medicine, 21st Edition, pp. 8436, 8440, 8462
This is a shared conversation. Sign in to Orris to start your own chat.