CFTR
Reading File
search_medical_text
CFTR gene protein function cystic fibrosis
search_medical_text
CFTR mutations classes pathophysiology diagnosis treatment
search_medical_image
CFTR protein structure cystic fibrosis
GradeResponse
GradeResponse
GradeResponse
CFTR — Cystic Fibrosis Transmembrane Conductance Regulator
Gene & Protein
CFTR is located on chromosome 7q31.2 and encodes a ~1,480 amino acid membrane glycoprotein that belongs to the ABC (ATP-binding cassette) transporter superfamily. It functions primarily as a cAMP-regulated anion (Cl⁻/HCO₃⁻) channel on the apical (luminal) plasma membrane of epithelial cells, regulating the volume and composition of exocrine secretions (Harrison's, p. 8006).
Protein domains:
| Domain | Function |
|---|---|
| 2× Membrane-spanning domains (MSD1, MSD2) | Form the ion channel pore |
| 2× Nucleotide-binding domains (NBD1, NBD2) | Bind/hydrolyze ATP; gate the channel |
| Regulatory (R) domain | PKA phosphorylation site; required for activation |
Mutation Classes
Six functional classes of CFTR mutations are recognized, each disrupting the protein at a different step:
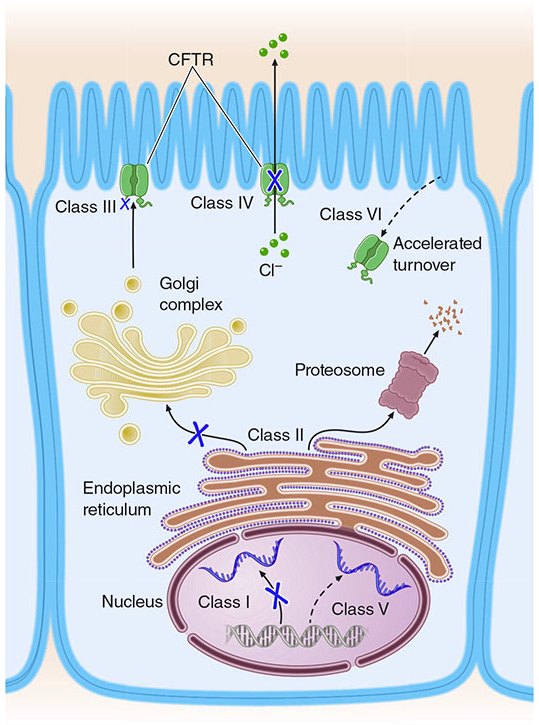
| Class | Defect | Example Mutation | Result |
|---|---|---|---|
| I | No protein synthesis | G542X, W1282X (nonsense/frameshift) | Absent CFTR |
| II | Misfolding → proteasomal degradation | F508del (most common, ~70% of alleles) | No surface CFTR |
| III | Reaches membrane but gating defect | G551D | Non-functional channel |
| IV | Reduced conductance through pore | R117H | Partial function |
| V | Reduced transcription/splicing | 3849+10kbC→T | Low but functional CFTR |
| VI | Reduced stability at cell surface | 4326delTC | Accelerated degradation |
(Harrison's, p. 8014)
Pathophysiology in Cystic Fibrosis (CF)
CF is autosomal recessive — two defective CFTR alleles required. Loss of CFTR function leads to:
- Airways: Impaired Cl⁻/HCO₃⁻ secretion + hyperactive ENaC (Na⁺ absorption) → dehydrated, viscous mucus → mucus plugging, chronic bacterial infection (Pseudomonas aeruginosa, S. aureus) → bronchiectasis
- Pancreas: Ductal obstruction → exocrine insufficiency (malabsorption, fat-soluble vitamin deficiency), endocrine failure (CF-related diabetes, CFRD)
- Liver: Inspissated bile → focal biliary cirrhosis
- Sweat glands: Failure to reabsorb Cl⁻ → elevated sweat chloride (diagnostic hallmark, >60 mmol/L)
- Reproductive tract: Congenital bilateral absence of vas deferens (CBAVD) in males; reduced fertility in females
Diagnosis
| Test | Finding in CF |
|---|---|
| Newborn screen | Elevated immunoreactive trypsinogen (IRT), followed by DNA analysis |
| Sweat chloride | ≥60 mmol/L (diagnostic); 40–59 borderline |
| CFTR mutation analysis | Identifies pathogenic variants on both alleles |
| Nasal potential difference | Functional CFTR assay; used in borderline cases |
CFTR Modulator Therapy
Modern small-molecule modulators target specific defect classes:
| Drug Class | Mechanism | Agent(s) | Target Mutation(s) |
|---|---|---|---|
| Potentiators | Improve gating of surface CFTR | Ivacaftor (VX-770) | Class III (G551D, etc.) |
| Correctors | Rescue misfolded protein trafficking | Lumacaftor, Tezacaftor, Elexacaftor | Class II (F508del) |
| Combination | Corrector(s) + potentiator | Elexacaftor/tezacaftor/ivacaftor (ETI, Trikafta) | F508del (one or two alleles); most CF patients eligible |
| Read-through agents (investigational) | Suppress premature stop codons | Ataluren | Class I |
Elexacaftor/tezacaftor/ivacaftor (ETI) is now the standard of care for eligible patients (~90% of CF patients), producing ~10-point improvements in ppFEV₁ and dramatic reductions in pulmonary exacerbations.
Key Clinical Points
- F508del accounts for ~70% of mutant alleles worldwide; homozygous F508del is the most common CF genotype
- Disease severity correlates imperfectly with genotype — modifier genes and environment matter
- CFTR is also expressed in the gut, liver, reproductive tract, and sweat glands, explaining the multi-organ phenotype
- CBAVD without lung disease can be caused by milder CFTR variants (e.g., 5T allele) — CFTR testing is part of male infertility workup
- Pancreatic-sufficient patients (Classes IV/V) generally have better pulmonary prognosis and slower lung decline
CFTR
search_medical_text
CFTR cystic fibrosis clinical manifestations complications management
search_medical_text
CFTR modulator therapy ivacaftor elexacaftor tezacaftor outcomes
search_medical_image
cystic fibrosis lung pathology bronchiectasis
GradeResponse
GradeResponse
GradeResponse
It looks like you've queried "CFTR" twice. The previous response already covered CFTR comprehensively. Here's a focused recap — let me know if you'd like to go deeper on any specific aspect.
CFTR — Quick Reference
Cystic Fibrosis Transmembrane Conductance Regulator is a cAMP-activated Cl⁻/HCO₃⁻ anion channel on the apical surface of epithelial cells. Loss-of-function mutations (autosomal recessive) cause cystic fibrosis (CF).
Mutation Classes (Harrison's, p. 8014)
| Class | Defect | Prototype |
|---|---|---|
| I | No synthesis | G542X |
| II | Misfolding/degradation | F508del (~70% of alleles) |
| III | Gating failure at membrane | G551D |
| IV | Reduced conductance | R117H |
| V | Reduced transcripts | 3849+10kbC→T |
| VI | Accelerated turnover | 4326delTC |
End-Organ Effects of CFTR Dysfunction
- Lungs: Dehydrated mucus → chronic Pseudomonas infection → bronchiectasis (see below)
- Pancreas: Exocrine insufficiency, CFRD
- Liver: Focal biliary cirrhosis
- Sweat glands: Elevated sweat Cl⁻ (≥60 mmol/L — diagnostic)
- Male reproductive: CBAVD → infertility
CFTR Modulators (Harrison's, p. 8017)
| Agent(s) | Class | Mechanism | Eligible Genotype |
|---|---|---|---|
| Ivacaftor | Potentiator | Increases channel open probability | Class III (G551D, etc.) |
| Lumacaftor/tezacaftor + ivacaftor | Corrector + potentiator | Rescues F508del trafficking | Homozygous F508del |
| Elexacaftor/tezacaftor/ivacaftor (ETI, Trikafta) | Next-gen corrector + potentiator | Dual corrector mechanism + gating | ≥1 F508del allele; >90% of CF patients |
ETI is the current standard of care, yielding ~10-point ppFEV₁ gains and major reductions in exacerbations.
What aspect would you like to explore further — genetics, pulmonary management, non-pulmonary complications, modulators, or something else?
This is a shared conversation. Sign in to Orris to start your own chat.