Inclusion body myositis full details for md exams 15marks
Reading File
search_medical_text
inclusion body myositis pathology etiology clinical features diagnosis
search_medical_text
inclusion body myositis treatment management prognosis EMG muscle biopsy
search_medical_image
inclusion body myositis muscle biopsy rimmed vacuoles histology
GradeResponse
GradeResponse
GradeResponse
Here is a comprehensive, exam-oriented answer on Inclusion Body Myositis (IBM) structured for a 15-mark MD/postgraduate examination.
Inclusion Body Myositis (IBM)
Definition
Inclusion body myositis is the most common acquired inflammatory myopathy in patients over 50 years of age. It is a chronic, progressive, inflammatory and degenerative muscle disease characterized by selective muscle weakness, rimmed vacuoles on biopsy, and intracellular tubulofilamentous inclusions — and is notably refractory to immunosuppressive therapy, distinguishing it from other inflammatory myopathies.
Classification of Inflammatory Myopathies
| Feature | Polymyositis (PM) | Dermatomyositis (DM) | Inclusion Body Myositis (IBM) |
|---|---|---|---|
| Age of onset | Any age | Any age (bimodal) | >50 years |
| Sex | F > M | F > M | M > F (3:1) |
| Pattern of weakness | Proximal | Proximal | Proximal + Distal |
| Skin involvement | No | Yes | No |
| Dysphagia | Uncommon | Uncommon | Common (60–80%) |
| Response to steroids | Good | Good | Poor |
| Malignancy association | Low | High | Low |
Epidemiology
- Most common inflammatory myopathy in adults over 50
- Prevalence: ~50/million (rises with age)
- Male > Female (3:1)
- Predominantly affects Caucasians
- Sporadic IBM (sIBM) is the common form; hereditary IBM (hIBM) is rare
Etiology and Pathogenesis
IBM has a dual pathogenesis — both inflammatory (autoimmune) and degenerative components are operative:
1. Inflammatory / Autoimmune Component
- CD8+ cytotoxic T-lymphocytes invade non-necrotic muscle fibers (same as PM)
- T cells are clonally expanded and antigen-driven
- MHC class I upregulation on muscle fibers
- Autoantibody: Anti-cN1A (anti-cytosolic 5'-nucleotidase 1A) — present in ~33–76% of IBM patients; relatively specific
2. Degenerative Component
- Abnormal protein accumulation within muscle fibers (similar to neurodegeneration)
- Accumulation of: β-amyloid precursor protein (β-APP), phosphorylated tau, TDP-43, p62, ubiquitin
- Endoplasmic reticulum stress → unfolded protein response
- Mitochondrial dysfunction with cytochrome oxidase (COX)-negative fibers
- Autophagic vacuole formation → rimmed vacuoles
Pathology (Muscle Biopsy)
This is the gold standard for diagnosis.
Light Microscopy (H&E Stain):
- Rimmed vacuoles — sarcoplasmic vacuoles lined by granular material (most characteristic)
- Endomysial inflammation — CD8+ T cells invading non-necrotic fibers
- Ragged red fibers (modified Gomori trichrome)
- Eosinophilic cytoplasmic or intranuclear inclusions
Histochemical Stains:
- COX (Cytochrome Oxidase) stain → increased COX-negative (pale) fibers
- p62 / TDP-43 immunostaining → cytoplasmic inclusions stain positive (highly specific)
- Congo red stain → amyloid deposits within vacuoles
Electron Microscopy:
- Tubulofilamentous inclusions: 15–21 nm diameter — intranuclear or cytoplasmic — pathognomonic
- Paired helical filaments
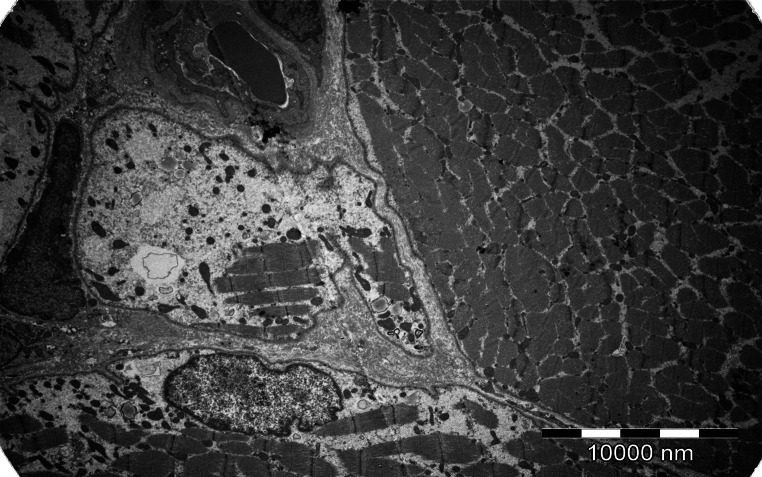
(Harrison's, 21st ed., p. 10284)
Clinical Features
Onset
- Insidious onset over months to years
- Often misdiagnosed as PM for years before correct diagnosis
Pattern of Weakness — Selective and Characteristic:
| Muscle Group | Finding |
|---|---|
| Finger flexors (FDP) | Early, selective weakness — inability to make a fist |
| Wrist flexors | Weakness > wrist extensors (reverse of most myopathies) |
| Quadriceps (knee extensors) | Early atrophy and weakness — difficulty rising from chair, frequent falls |
| Foot dorsiflexors | Foot drop |
| Pharyngeal muscles | Dysphagia — 60–80%; can be the presenting feature |
Key exam point: IBM causes asymmetric, distal + proximal weakness, with selective involvement of finger flexors and quadriceps — this pattern is virtually diagnostic.
Other Features:
- Atrophy of forearm flexors and quadriceps
- Hyporeflexia (reduced deep tendon reflexes)
- Mild or no muscle pain
- Falls are common due to quadriceps weakness
- No skin rash, no Raynaud's, no arthritis
- Rare systemic involvement (cardiac, pulmonary less common than PM/DM)
Investigations
1. Serum Creatine Kinase (CK)
- Mildly elevated — typically < 12× upper limit of normal (often only 2–5× ULN)
- May be normal in up to 20% of cases
- (Contrast: PM/DM — markedly elevated)
2. Autoantibodies
- Anti-cN1A (anti-NT5C1A) — present in ~33–76%; associated with more severe dysphagia
- MSA (myositis-specific antibodies like anti-Jo1) are typically absent
3. Electromyography (EMG)
- Mixed pattern (unique to IBM):
- Myopathic features: short-duration, low-amplitude, polyphasic potentials
- Neuropathic features: long-duration potentials, fibrillations
- This mixed pattern often leads to misdiagnosis as motor neuron disease
4. Muscle Biopsy
(Gold standard — see Pathology section above)
5. MRI of Muscles
- Early involvement of vastus lateralis and vastus medialis with relative sparing of rectus femoris — characteristic pattern on thigh MRI (Harrison's, 21st ed., p. 12782)
- MRI is more sensitive than EMG or clinical exam in early disease
- Helps guide biopsy site selection
6. Other Labs
- ESR, CRP: mildly elevated or normal
- ANA: may be weakly positive
- CBC, metabolic panel: to exclude systemic causes
Diagnostic Criteria
European Neuromuscular Centre (ENMC) 2011 Criteria — Three levels:
| Category | Requirements |
|---|---|
| Clinico-pathologically defined IBM | Age >45, duration >12 months, weakness of finger flexors OR knee extensors; PLUS biopsy showing endomysial inflammation + rimmed vacuoles + protein inclusions (p62/TDP-43) OR tubulofilamentous inclusions on EM |
| Clinically defined IBM | Clinical features (above) + biopsy showing endomysial inflammation + rimmed vacuoles (inclusions not required) |
| Probable IBM | Clinical features but biopsy only shows endomysial inflammation without rimmed vacuoles |
Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Polymyositis | Proximal only, responds to steroids, no rimmed vacuoles |
| Limb-girdle muscular dystrophy | Genetic, no inflammation on biopsy |
| Motor neuron disease (ALS) | Upper + lower motor neuron signs, EMG shows pure denervation |
| Myasthenia gravis | Fatigable weakness, positive AChR antibodies, responds to treatment |
| Oculopharyngeal muscular dystrophy | Ptosis + dysphagia, PABPN1 mutation, GCG repeat |
Treatment
Immunosuppressive Therapy
Critical exam point: IBM is largely refractory to all immunosuppressive therapies — unlike PM and DM.
| Drug | Evidence |
|---|---|
| Corticosteroids | No benefit in controlled trials; may worsen muscle function |
| Methotrexate, Azathioprine | No proven benefit |
| IVIG | Transient, modest benefit in dysphagia; not sustained |
| Alemtuzumab, anti-T cell therapies | Experimental, no proven benefit |
| Bimagrumab (anti-ActRII) | Phase II trials — shows promise for muscle mass |
| Rapamycin (sirolimus) | Ongoing trials targeting autophagy pathway |
Supportive / Rehabilitative (Most Important)
- Physiotherapy — resistance exercises to slow functional decline; eccentric strengthening of quadriceps
- Occupational therapy — adaptive devices (grip aids, jar openers)
- Speech therapy + dysphagia management — modified diet, cricopharyngeal myotomy or dilation for severe dysphagia
- Orthoses — ankle-foot orthoses (AFO) for foot drop
- Fall prevention — walking aids, home modifications
Prognosis
- Slowly but relentlessly progressive — most patients require a walking aid within 10–15 years
- No spontaneous remissions
- Major morbidity:
- Loss of ambulation (wheelchair dependence)
- Aspiration pneumonia from dysphagia (leading cause of death)
- Respiratory failure (late)
- Life expectancy minimally reduced if complications (aspiration, falls) are managed
- No increased risk of malignancy (unlike DM)
Summary: High-Yield Exam Points
| Feature | Key Point |
|---|---|
| Most common myopathy in | Adults >50 years |
| Sex predilection | Male > Female (3:1) |
| Pattern of weakness | Finger flexors + Quadriceps (proximal + distal, often asymmetric) |
| Dysphagia | 60–80%; can be presenting symptom |
| CK | Mildly elevated or normal |
| EMG | Mixed myopathic + neuropathic pattern |
| Biopsy hallmark | Rimmed vacuoles + CD8+ T cell invasion + tubulofilamentous inclusions (15–21 nm) |
| Pathognomonic EM finding | Tubulofilamentous inclusions (15–21 nm) |
| Specific autoantibody | Anti-cN1A (anti-NT5C1A) |
| MRI thigh finding | Vastus lateralis/medialis involvement, rectus femoris spared |
| Response to steroids | Poor / None |
| Treatment of choice | Physiotherapy + supportive care |
| Leading cause of death | Aspiration pneumonia |
Sources: Harrison's Principles of Internal Medicine, 21st Edition (pp. 10284, 12782); ENMC IBM Diagnostic Criteria 2011; Neurology clinical guidelines.
This is a shared conversation. Sign in to Orris to start your own chat.