hernias of the anterior abdominal wall, inguinal hernias-D Tomorrow class topic can you make detiald ppt to download
anterior abdominal wall hernias anatomy classification types
inguinal hernia anatomy pathophysiology direct indirect femoral repair surgery
inguinal hernia anatomy direct indirect diagram
- Title slide
- Learning objectives
- Anterior abdominal wall hernias overview
- Classification (primary ventral, incisional, parastomal, etc.)
- Inguinal canal anatomy
- Direct vs indirect inguinal hernia
- Femoral vs inguinal comparison
- Clinical features
- Examination findings
- Investigations
- Complications
- Management principles
- Surgical options (open vs lap/robotic)
- Post-op and recurrence prevention
- Case discussion slide
- Summary + viva questions
- Bailey & Love, 28th ed, p.1087 (direct/indirect/femoral relation)
- your level (MBBS / nursing / surgery residency)
- desired length (30 min / 45 min / 60 min)
Mbbs pedatric department
inguinal hernia children pediatric pathophysiology processus vaginalis congenital
pediatric inguinal hernia clinical features diagnosis management surgery children
anterior abdominal wall hernia types umbilical epigastric pediatric
Hernias of the Anterior Abdominal Wall & Inguinal Hernia (Pediatrics Focus)
Slide 1: Title
MBBS Pediatrics Teaching Session
Presenter: __________
Date: __________
Slide 2: Learning Objectives
- Define anterior abdominal wall hernias
- Classify common pediatric abdominal wall hernias
- Describe embryology and anatomy relevant to pediatric inguinal hernia
- Differentiate direct vs indirect inguinal hernia (conceptual and clinical)
- Recognize presentation, complications, and red flags
- Outline investigations and treatment principles in children
- Understand basics of herniotomy and perioperative care
Slide 3: Definition & Scope
- Ventral (anterior abdominal wall) hernia: protrusion through defects in anterior abdominal wall
- Includes: umbilical, epigastric, incisional, parastomal (less common in children)
- Groin hernias (inguinal/femoral) are discussed separately in many classifications
Slide 4: Pediatric Relevance
- Hernias are common pediatric surgical referrals
- Most pediatric inguinal hernias are congenital
- Early diagnosis prevents incarceration/strangulation
- Important for MBBS students to identify urgent vs non-urgent cases
Slide 5: Embryology of Pediatric Inguinal Hernia
- Testis descends with peritoneal outpouching (processus vaginalis)
- Normally processus vaginalis obliterates
- Failure of closure leads to patent processus vaginalis and indirect inguinal hernia
Slide 6: Inguinal Canal Anatomy (Exam Slide)
- Deep inguinal ring
- Superficial inguinal ring
- Inferior epigastric vessels
- Spermatic cord/round ligament
- Hesselbach triangle (for direct hernia concept)
Slide 7: Direct vs Indirect vs Femoral (Comparison)
| Feature | Indirect Inguinal | Direct Inguinal | Femoral |
|---|---|---|---|
| Relation to inferior epigastric vessels | Lateral | Medial | Below inguinal ligament |
| Common age group | Children/young | Older adults | More in adult females |
| Pathogenesis | Patent processus vaginalis | Posterior wall weakness | Femoral canal defect |
| Pediatric frequency | Very common | Rare | Rare |
Slide 8: Types of Pediatric Anterior Abdominal Wall Hernias
- Umbilical hernia
- Epigastric hernia
- Incisional hernia (post-surgical)
- Rare ventral defects depending on prior surgeries/congenital disorders
Slide 9: Umbilical Hernia in Children
- Common in infants
- More in prematurity/low birth weight
- Usually painless, reducible swelling at umbilicus
- Many close spontaneously by 2-5 years
- Surgery if persistent/large/complicated
Slide 10: Epigastric Hernia
- Midline defect between xiphoid and umbilicus
- Protrusion of preperitoneal fat ± peritoneal sac
- Small, localized swelling, sometimes painful
- Usually elective repair if symptomatic
Slide 11: Clinical Presentation of Pediatric Inguinal Hernia
- Intermittent groin/scrotal swelling
- Swelling increases on crying/coughing/straining
- Reducible swelling with normal child between episodes
- Parents often report “comes and goes”
Slide 12: Examination
- Inspect both groins and scrotum in standing/crying child (if possible)
- Check reducibility
- Assess contralateral side
- Distinguish from:
- Hydrocele
- Undescended testis
- Inguinal lymphadenopathy
- Femoral hernia (rare)
Slide 13: Red Flags (Emergency)
- Irreducible painful swelling
- Persistent crying/irritability
- Vomiting, abdominal distension
- Skin erythema/tenderness over swelling
- Features of bowel obstruction
Slide 14: Complications
- Incarceration
- Strangulation
- Bowel obstruction
- Testicular compromise (rare but serious)
- Recurrence (post repair, uncommon with proper technique)
Slide 15: Investigations
- Mainly clinical diagnosis
- Ultrasound only in selected uncertain cases
- Pre-op routine pediatric anesthesia fitness workup
Slide 16: Management Principles
Inguinal hernia in children:
- Definitive treatment is surgery (herniotomy)
- Do not delay unnecessarily after diagnosis
- Earlier surgery in infants due to incarceration risk
Umbilical hernia:
- Observe initially (most spontaneous closure)
- Operate if persistent/large/complicated
Slide 17: Surgical Overview (MBBS Level)
- Pediatric inguinal herniotomy:
- Groin incision
- Identify sac
- High ligation of sac at internal ring
- Preserve vas and vessels
- Mesh usually not required in routine pediatric indirect hernia
- Laparoscopic options increasingly used
Slide 18: Postoperative Care
- Day-care surgery in many cases
- Pain control and wound care
- Parents advised on:
- Fever, swelling, redness, persistent pain
- Vomiting or recurrence of groin bulge
- Follow-up for wound and recurrence check
Slide 19: Special Pediatric Points
- Premature infants: higher risk and peri-anesthetic considerations
- Bilateral hernia possibility in infants
- Associated conditions: connective tissue disorders, raised intra-abdominal pressure states
- Counsel parents about urgency if swelling becomes painful/irreducible
Slide 20: Case-Based Discussion
Questions:
- Most likely diagnosis?
- Mechanism?
- Immediate management plan?
- What advice to parents for emergency return?
Slide 21: Viva / Short Questions
- Define hernia and differentiate reducible vs incarcerated
- Why are pediatric inguinal hernias mostly indirect?
- Difference between hydrocele and inguinal hernia
- Indications for surgery in umbilical hernia
- Complications of delayed treatment
Slide 22: Key Take-Home Messages
- Pediatric inguinal hernia is usually congenital (patent processus vaginalis)
- Clinical diagnosis is usually sufficient
- Early elective repair prevents incarceration/strangulation
- Umbilical hernia often closes spontaneously; selected cases need surgery
- Always educate parents about red-flag symptoms
Slide 23: References
- Bailey & Love’s Short Practice of Surgery, 28th Edition
- Ventral hernia classification (p.1093)
- Congenital origin/processus vaginalis (p.1080)
- Inguinal hernia summary and management approach (p.1086)
- Groin hernia anatomy diagram (p.1087)
Suggested Images to Insert
- Direct vs indirect vs femoral anatomy diagram
Source: Bailey & Love 28th ed, p.1087 - Pediatric epigastric hernia clinical pre/post image (if your department permits clinical photos)
- a ready 5-minute speaker script per slide, or
- a short 12-slide version for quick presentation, or
- a final .pptx-building template text (copy once into PowerPoint Designer).
ауруы/Malformations of the digestive tract. Congenital intestinal obstruction. Atresia of the anus and rectum. Hirschsprung's disease-PD-D Everything according to pedia surgery department
Hirschsprung disease aganglionic megacolon barium enema radiology
intestinal atresia types classification congenital duodenal small bowel pediatric
anorectal malformation imperforate anus classification types pediatric surgery
📋 Slide Structure
| Slides | Content |
|---|---|
| 1–2 | Title + Learning Objectives |
| 3–4 | Embryology + Classification overview |
| 5 | General signs of neonatal obstruction |
| 6–8 | Duodenal Atresia (pathogenesis, double bubble, surgery) |
| 9–11 | Jejunoileal Atresia (Grosfeld classification + intraoperative images) |
| 12–17 | Hirschsprung's Disease (full coverage: genetics → diagnosis → surgery) |
| 18–23 | Anorectal Malformations (ARM) + VACTERL + Management |
| 24 | Master comparison table |
| 25–26 | Case discussions + Viva questions |
| 27–28 | Take-home messages + References |
🖼️ Images Embedded
- Double bubble sign X-ray (duodenal atresia)
- Grosfeld classification diagram (jejunoileal atresia)
- Intraoperative types I–IV
- Hirschsprung's barium enema transition zone
- Hirschsprung's barium enema + histopathology (H&E)
- Female rectovestibular fistula (clinical)
- Male imperforate anus (clinical)
- Invertogram for ARM
I don't need slides i need full details of topic for class discussion
MALFORMATIONS OF THE DIGESTIVE TRACT
Congenital Intestinal Obstruction | Atresia of the Anus & Rectum | Hirschsprung's Disease
MBBS Pediatric Surgery — Full Class Discussion Notes
PART 1: EMBRYOLOGICAL BASIS (Foundation for All Topics)
- Foregut → esophagus, stomach, duodenum (proximal), liver, pancreas, bile ducts
- Midgut → duodenum (distal) to mid-transverse colon; undergoes herniation into umbilical cord and returns at week 10 with 270° counterclockwise rotation
- Hindgut → distal transverse colon → rectum → upper anal canal
- Anterior: urogenital sinus
- Posterior: anorectal canal
PART 2: CONGENITAL INTESTINAL OBSTRUCTION
2.1 General Principles
- Bilious (green) vomiting — the single most important sign; any bile-stained vomit in a neonate must be treated as bowel obstruction until proven otherwise
- Abdominal distension — the more distal the obstruction, the greater the distension
- Failure to pass meconium within the first 24–48 hours of life
- Polyhydramnios detected on prenatal ultrasound (swallowed amniotic fluid cannot be absorbed past the obstruction → accumulates)
- Prenatal USS showing dilated bowel loops or double bubble
- Associated anomalies: Down syndrome, cardiac defects, vertebral/renal anomalies
- Obstruction proximal to the Ampulla of Vater → non-bilious vomiting (gastric outlet, pyloric, rare proximal duodenal)
- Obstruction distal to the Ampulla of Vater → bilious vomiting (85% of duodenal atresia, all jejunoileal atresia)
2.2 Duodenal Atresia and Stenosis
Incidence
Pathogenesis
Anatomical Variants (from least to most severe)
- Duodenal stenosis — partial narrowing; incomplete lumen
- Mucosal web (windsock deformity) — intact muscle wall, only the mucosa forms an obstructing membrane; the web can bulge distally giving a "windsock" appearance — can cause delayed diagnosis if incomplete
- Two ends separated by a fibrous cord — complete obstruction, mesentery partially intact
- Complete separation with a gap — most severe, no continuity
Location
- 85% distal to the Ampulla of Vater → bile enters the proximal duodenum → bilious vomiting
- 15% proximal to ampulla → non-bilious vomiting
Associated Conditions (very important for MBBS — frequently tested)
- Down syndrome (Trisomy 21) — strongest association; ~30% of duodenal atresia cases have Down syndrome
- Prematurity
- Maternal polyhydramnios
- Intestinal malrotation
- Annular pancreas (pancreatic tissue wraps around the duodenum compressing it)
- Preduodenal portal vein (portal vein runs anterior to duodenum)
- Biliary atresia
- Cardiac anomalies, renal anomalies
- Esophageal atresia
- Other anorectal malformations
Clinical Features
- Prenatal: polyhydramnios on USS; dilated stomach + duodenum on fetal USS
- Postnatal: bilious vomiting from birth (within hours)
- Abdomen is scaphoid (flat/sunken) — because distal bowel contains no air
- Failure to pass meconium is variable (meconium may be passed as rectum/colon can develop normally)
Investigations
- Gas bubble 1 = dilated stomach
- Gas bubble 2 = dilated proximal duodenum
- Complete absence of distal gas = complete atresia
- If distal gas is present → incomplete obstruction (stenosis or mucosal web) → must do upper GI contrast study to:
- Confirm the diagnosis
- Exclude malrotation with midgut volvulus (surgical emergency that must not be missed)
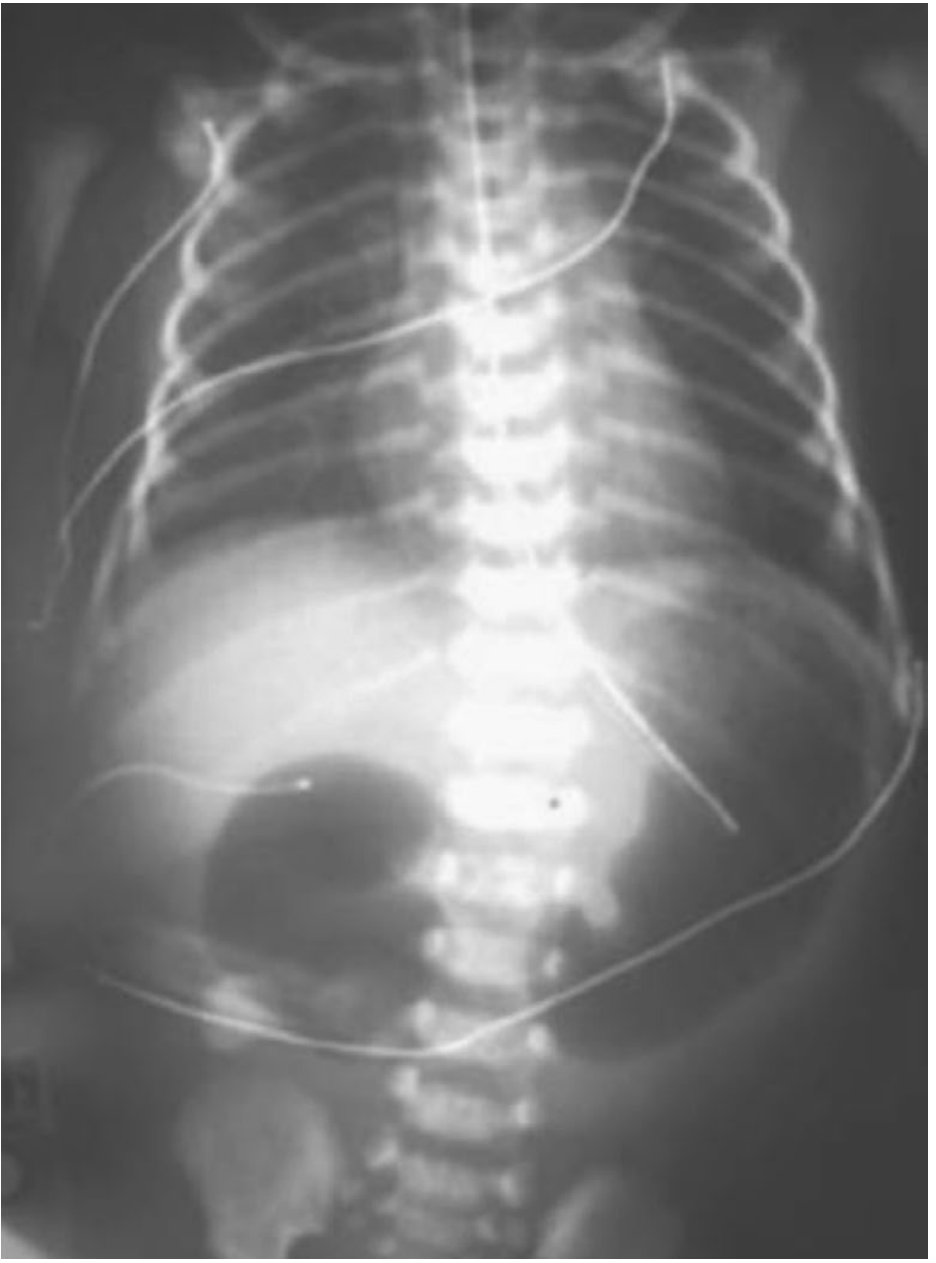
Management
- IV access, NGT decompression, fluid resuscitation
- Correct electrolytes (vomiting causes hypochloraemic metabolic alkalosis)
- Echocardiogram to exclude cardiac anomalies before surgery
- Duodenoduodenostomy — bypass of the obstruction:
- Side-to-side anastomosis, OR
- Diamond-shaped anastomosis (proximal transverse to distal longitudinal) — reduces anastomotic stricture
- Laparoscopic approach increasingly used with excellent outcomes
- If proximal duodenum is markedly dilated → tapering duodenoplasty to reduce dysmotility and stasis
- In mucosal web → web is fenestrated or excised transduodenally (caution: avoid ampulla injury)
2.3 Jejunoileal Atresia
Incidence
Pathogenesis
- Why it is associated with cystic fibrosis (meconium ileus causes volvulus → vascular insult)
- Why it is NOT strongly associated with Down syndrome or other chromosomal anomalies
- Why multiple atresias can occur (multiple ischemic events)
Grosfeld Classification (5 Types — exam essential)
| Type | Anatomy | Notes |
|---|---|---|
| Type I | Mucosal web/diaphragm; bowel wall and mesentery intact | Mildest; may be missed on inspection |
| Type II | Two blind ends joined by fibrous cord; mesentery intact | Clear discontinuity |
| Type IIIa | Complete separation of blind ends; V-shaped mesenteric gap | Most common type |
| Type IIIb | Apple peel / Christmas tree deformity — extensive mesenteric gap; distal bowel spirals around a single blood vessel (marginal artery of ileocolic) | Associated with short gut syndrome; very short bowel remnant |
| Type IV | Multiple atresias — "string of sausages" appearance; 10–15% of cases | Must evaluate entire bowel intraoperatively |
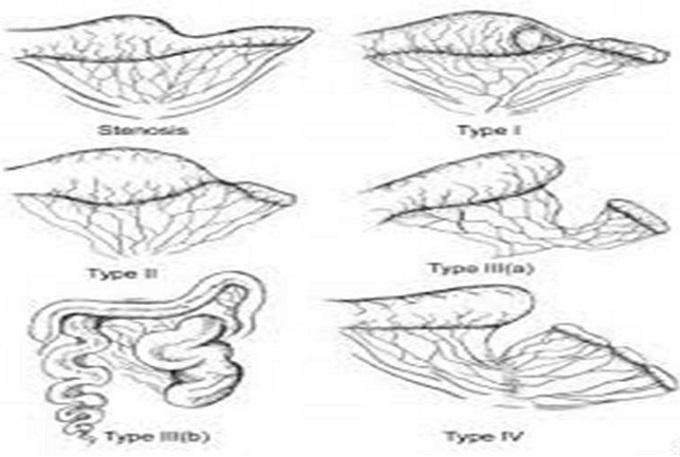
Clinical Features
- Proximal (jejunal) atresia: Prominent bilious vomiting, less distension
- Distal (ileal) atresia: Greater abdominal distension, multiple dilated loops on X-ray
- Failure to pass meconium
- On X-ray: dilated loops with air-fluid levels; microcolon on contrast enema (colon unused in utero → very small caliber)
Investigations
- Plain X-ray: multiple dilated loops with air-fluid levels; degree of distension depends on level
- Contrast enema: shows microcolon (unused colon); rarely needed to diagnose but useful to exclude Hirschsprung's or other diagnoses
- Note: Do NOT delay surgery for extensive investigations
Management
- Resect the atretic segment
- End-to-end or end-to-oblique anastomosis
- Critical intraoperative step: inject saline via a soft red rubber catheter into distal bowel to check for additional atresias (multiple atresias in 10–15%)
- Type IIIb (apple peel): high risk of short gut syndrome; conserve as much bowel as possible
- Post-op TPN often required until anastomosis and bowel function recover
2.4 Other Causes of Congenital Intestinal Obstruction (Brief Overview)
- Failure of normal 270° counterclockwise rotation of the midgut at week 10
- Midgut attached by narrow pedicle → prone to volvulus
- Ladd's bands cross the duodenum → extrinsic obstruction
- Midgut volvulus = pediatric surgical emergency: ischemia of entire midgut within hours
- Presents as sudden bilious vomiting in an otherwise well child; X-ray may show "double bubble" or non-specific dilation
- Treatment: Ladd's procedure (detorsion, divide Ladd's bands, appendectomy, widen mesenteric base)
- Impacted viscid meconium in distal ileum → obstruction
- 95% associated with cystic fibrosis (abnormal mucus)
- X-ray: ground-glass appearance (Neuhauser sign), no air-fluid levels (meconium too thick to layer)
- Uncomplicated: Gastrografin enema (hyperosmolar — draws water, loosens meconium)
- Complicated (perforation, volvulus, atresia): surgery
PART 3: ATRESIA OF THE ANUS AND RECTUM (ANORECTAL MALFORMATIONS)
3.1 Introduction and Incidence
- Incidence: 1 in 5,000 live births
- Sex predominance: Male 58%, Female 42%
- The spectrum ranges from minor perineal groove with normal anus → severe cloaca (single opening for urinary, reproductive, and digestive tracts)
3.2 Embryology and Pathogenesis
- At week 5–6: The urorectal septum descends caudally, dividing the cloaca into:
- Anterior compartment → urogenital sinus (bladder, urethra, vagina)
- Posterior compartment → anorectal canal
- At week 7–8: The cloacal membrane breaks down; the anal membrane resorbs to create the anal opening
- Cloacal folds extend from the genital tubercle to the anus → fuse to form the perineal body between the anal and urogenital openings
- Failure of urorectal septum to descend properly → fistula between rectum and urinary tract (in males) or introitus (in females)
- Failure of the anal membrane to resorb → anal membrane, anal stenosis
- Breakdown of the cloacal membrane at wrong site → external anal opening displaced anterior to the external sphincter (perineal fistula)
- Complete failure of anorectal canal development → imperforate anus without fistula
3.3 Classification
In MALES:
| Type | Description |
|---|---|
| Cutaneous (perineal fistula) | Rectum opens onto perineal skin anterior to sphincter — lowest/mildest |
| Rectobulbar urethral fistula | Rectum communicates with the bulbar (posterior) urethra |
| Rectoprostatic urethral fistula | Rectum communicates with prostatic urethra — more proximal |
| Recto-bladder neck fistula | Rectum communicates with bladder neck — highest/most severe in males |
| Imperforate anus without fistula | Blind-ending rectal pouch, no fistula |
| Rectal atresia | Lumen of rectum interrupted; upper rectum dilated, lower = small anal canal |
In FEMALES:
| Type | Description |
|---|---|
| Cutaneous (perineal fistula) | Opens onto perineal skin |
| Vestibular fistula | Most common in females — rectum opens into the vaginal vestibule |
| Imperforate anus without fistula | Blind pouch |
| Rectal atresia | — |
| Cloaca | Rectum, vagina, and urethra fuse into a single common channel opening at the perineum — the most complex ARM |
3.4 Clinical Features and Diagnosis
- No visible anal opening → obvious imperforate anus
- Anal opening present but abnormally positioned or stenotic
- Meconium visible at perineal skin, at the base of the scrotum (males), or in the vaginal vestibule (females)
- Meconium in urine (rectourinary fistula) — pathognomonic of rectourethral fistula
- Flat perineum, poor sphincter complex (suggests high lesion)
- Well-formed buttocks with good midline groove = low lesion (good prognosis for continence)
3.5 Investigations
- Infant held upside down for 3–5 minutes, lateral X-ray taken
- Gas rises to the most distal point of the rectal pouch
- Radiopaque marker placed on the skin at the expected anal site
- Distance from gas bubble to marker determines high vs. low
- Now largely replaced but still taught and tested
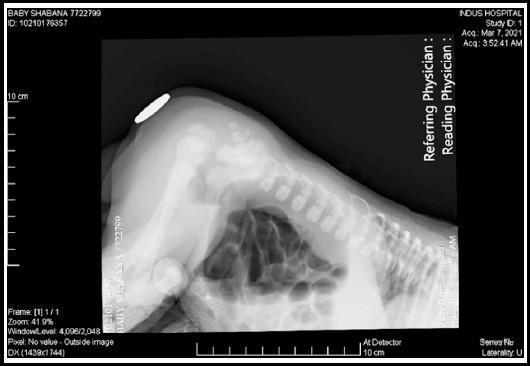
- Current gold standard to define fistula anatomy before definitive repair
- Contrast instilled into distal limb of colostomy under pressure → outlines fistula location
3.6 VACTERL Association (Must Know)
| Letter | Anomaly |
|---|---|
| V | Vertebral defects (sacral agenesis, hemivertebra) |
| A | Anorectal malformations |
| C | Cardiac defects (VSD most common) |
| TE | Tracheo-Esophageal fistula ± esophageal atresia |
| R | Renal anomalies (renal agenesis, horseshoe kidney, VUR) |
| L | Limb defects (radial aplasia, polydactyly) |
3.7 Management
Low lesions (perineal/vestibular fistula visible):
- Neonatal period: Perform anorectoplasty or fistula dilation in selected low cases
- Definitive repair: PSARP (Posterior Sagittal Anorectoplasty) by Peña and De Vries — the standard operation worldwide
- Patient in prone jack-knife position
- Posterior sagittal incision from coccyx to planned anal site
- Identify and mobilize the rectal pouch under direct vision
- Divide fistula, reconstruct sphincter mechanism, place rectum within the sphincter complex
- Highly precise — allows identification and preservation of sphincter muscles
- No colostomy needed in well-selected low lesions
High/intermediate lesions (no visible fistula, or proximal fistula):
- Stage 1 — Neonatal: Divided sigmoid loop colostomy (decompression; allows distal colostogram later)
- Stage 2 — Age 3–6 months: Definitive repair — PSARP ± laparotomy/laparoscopy for very high lesions
- Stage 3 — After confirmed healing: Colostomy closure
Cloaca:
- Always requires colostomy first
- Most complex repair — simultaneous reconstruction of rectum, vagina, urethra
- Common channel >3 cm → requires vaginal replacement/reconstruction
- Urological involvement often necessary
3.8 Prognosis and Long-Term Outcomes
- Type of malformation (low > high)
- Sacral development (sacral ratio)
- Quality of sphincter muscles
- Tethered cord (requires neurosurgical release before bowel repair)
PART 4: HIRSCHSPRUNG'S DISEASE (Congenital Aganglionic Megacolon)
4.1 Definition
4.2 Epidemiology
- Incidence: 1 in 5,000 live births
- Male:Female = 4:1 (short segment disease; this ratio narrows to 2:1 in long segment and 1:1 in total colonic aganglionosis)
- 3–5% have Down syndrome (Trisomy 21) — and conversely, ~2% of Down syndrome patients have Hirschsprung's
- Genetics: Abnormal locus on chromosome 10 associated with the RET proto-oncogene (a tyrosine kinase receptor involved in neural crest cell migration and survival). Also: GDNF (glial cell-derived neurotrophic factor), GFRα-1 coreceptor mutations identified
- Positive family history increases risk
- Associations: Down syndrome, MEN2A, congenital central hypoventilation syndrome (Ondine's curse)
4.3 Pathophysiology (Most Important for Class Discussion)
- No parasympathetic ganglion cells → absent inhibitory relaxation of the distal bowel and internal anal sphincter
- Tonic contraction of the aganglionic segment → functional obstruction
- Peristaltic waves cannot propagate across the aganglionic zone
- Proximal ganglionic bowel becomes distended with gas and stool → megacolon
- Paradox: The NARROW segment is the ABNORMAL one (aganglionic); the DILATED segment is the NORMAL one (ganglionic but obstructed proximally)
| Segment | % of cases |
|---|---|
| Rectosigmoid | ~80% (short segment — classic) |
| Splenic flexure to transverse colon | ~17% (long segment) |
| Entire colon ± small bowel | ~8% (total colonic aganglionosis — most severe) |
4.4 Clinical Features
Neonatal Presentation (>90% of cases):
- Failure to pass meconium within the first 24–48 hours of life — the hallmark sign
- Abdominal distension — progressively worsening
- Bilious vomiting
- Rectal examination → anal canal feels tight; withdrawal of finger → explosive release of gas and stool ("squirt sign") — characteristic but not pathognomonic
Later Presentation (missed or short-segment cases):
- Chronic constipation since birth (parent may report "never had a normal bowel movement")
- Failure to thrive, growth retardation
- Chronic abdominal distension
- Ribbon-like stools
Hirschsprung-Associated Enterocolitis (HAEC) — THE EMERGENCY:
- Leading cause of death in untreated Hirschsprung's disease
- Can occur before OR after surgical repair
- Pathogenesis: stasis → bacterial overgrowth → mucosal inflammation → enterocolitis
- Clinical features: Explosive foul-smelling diarrhea alternating with obstipation, abdominal distension, fever, hematochezia, peritonitis, septic shock, death
- Treatment: IV fluids, antibiotics (metronidazole + broad-spectrum), rectal washouts; emergency colostomy if deteriorating
4.5 Diagnosis
1. Contrast Enema (first investigation in newborns):
- Shows narrow aganglionic distal rectum → transition zone → dilated normal proximal sigmoid
- Reversed rectosigmoid ratio = rectum narrower than sigmoid (normally rectum > sigmoid)
- Contrast retention at 24 hours (failure to evacuate all contrast) strongly suggests Hirschsprung's
- Also useful to exclude: meconium plug syndrome, small left colon syndrome, intestinal atresia
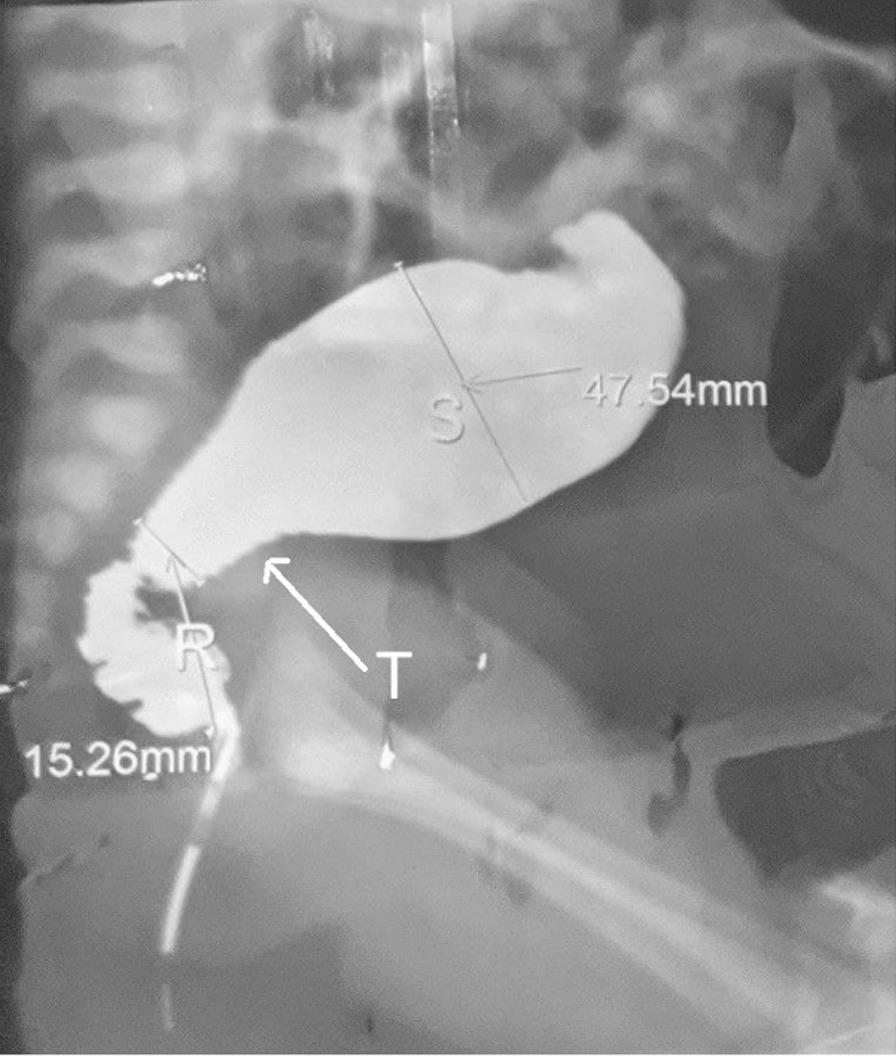
2. Anorectal Manometry (in toddlers and older children):
- Rectal balloon distension → normal response = internal sphincter RELAXES (recto-anal inhibitory reflex, RAIR)
- In Hirschsprung's: RAIR is absent — internal sphincter fails to relax
- Sensitivity ~83–93%; not reliable in neonates
3. Rectal Biopsy — GOLD STANDARD:
- Suction rectal biopsy — bedside in neonates, no anesthesia required; uses a suction biopsy kit
- Biopsy site: ≥5 mm to 1 cm above the dentate line (anoderm is physiologically hypoganglionic → false positive if too low)
- In infants: take 2+ specimens, 1 cm apart in posterior rectum
- In older children: full-thickness biopsy under GA (thicker mucosa not amenable to suction technique)
- Absent ganglion cells in the submucosal and myenteric plexus
- Hypertrophied nerve trunks (thickened, acetylcholinesterase-positive nerve fibers)
- Robust acetylcholinesterase (AChE) staining — positive (normally negative in ganglionic bowel)
- Calretinin immunostaining — absent in aganglionic bowel (now a standard adjunct; calretinin marks normal ganglion cells and small nerve fibers)
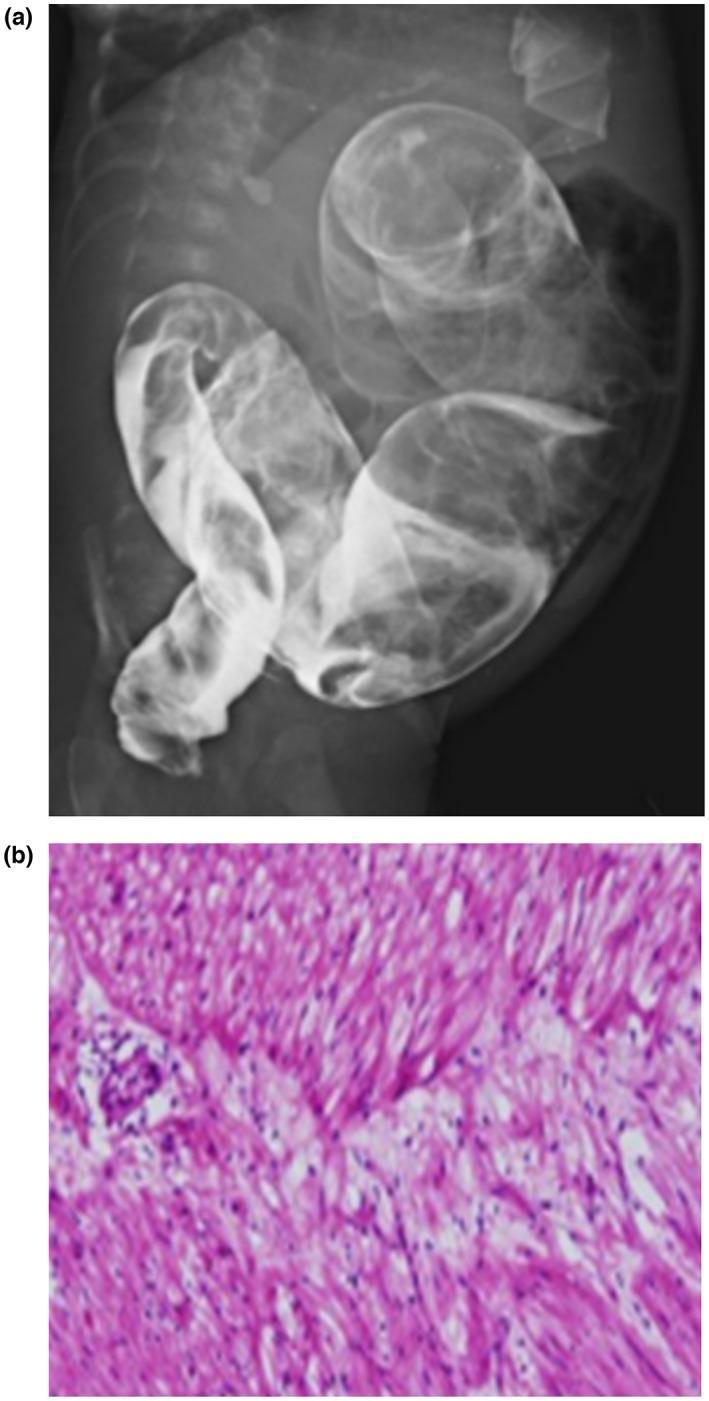
4.6 Management
Pre-operative Stabilization:
- Daily rectal irrigations with warm normal saline via a soft red rubber catheter
- Purpose: decompress the obstructed proximal bowel, reduce bacterial load, prevent enterocolitis
- Performed 1–2× daily, using as much saline as needed until the effluent returns clear
- If irrigations are not effective or not possible (long-segment disease, family unable to perform, comorbidities) → leveling colostomy — sigmoid divided at the level of confirmed ganglionic bowel (confirmed with intraoperative frozen section biopsies)
Definitive Surgical Treatment — Pull-Through Procedures:
- Full-thickness dissection of the rectum from surrounding sphincter mechanism
- Rectum pulled through; colo-anal anastomosis 5 mm above dentate line
- Risk: injury to pelvic autonomic nerves, bladder/sexual dysfunction
- Leaves the muscular cuff of the aganglionic rectum in place
- Strips out rectal mucosa; ganglionic bowel pulled through the muscular cuff
- Reduces risk of pelvic nerve injury
- Complication: retained aganglionic muscle cuff → residual obstruction (Soave cuff)
- Ganglionic colon brought down behind the aganglionic rectum
- Side-to-side colo-rectal anastomosis using a stapler
- Aganglionic anterior wall of rectum remains (can cause "spur" obstruction if staple line incomplete)
- Minimally invasive versions of the above (especially laparoscopic Soave)
- Increasingly used, especially in specialist centers
- Associated with shorter hospital stay, less adhesion risk, excellent outcomes
4.7 Post-operative Care and Complications
- Anastomotic leak
- Wound infection
- Temporary constipation as bowel adjusts
- Post-operative enterocolitis — can still occur after surgery; managed with rectal washouts and antibiotics
- Obstructive symptoms — due to residual aganglionic segment, anastomotic stricture, or Soave cuff
- Fecal incontinence — especially in long-segment disease or after complex repairs
- Constipation relapse — requires investigation for residual aganglionosis or stricture
4.8 Differential Diagnosis
| Condition | Key Distinguishing Feature |
|---|---|
| Meconium plug syndrome | Contrast enema therapeutic; resolves with enema; no biopsy change |
| Small left colon syndrome | Infant of diabetic mother; resolves spontaneously |
| Hypothyroidism | Constipation but thyroid function abnormal; no transition zone |
| Intestinal atresia | Different anatomy on contrast study; no megacolon |
PART 5: MASTER COMPARISON TABLE (Class Discussion Summary)
| Feature | Duodenal Atresia | Jejunoileal Atresia | Hirschsprung's Disease | Anorectal Malformation |
|---|---|---|---|---|
| Incidence | 1:5,000–10,000 | 1:2,000 | 1:5,000 | 1:5,000 |
| Pathogenesis | Failed recanalization | Vascular accident in utero | Neural crest migration arrest | Urorectal septum/cloacal failure |
| Key clinical sign | Bilious vomiting from birth | Bilious vomiting + distension + no meconium | No meconium + constipation ± megacolon | No anal opening on perineal exam |
| Key radiology | Double bubble sign (X-ray) | Dilated loops + microcolon | Transition zone on contrast enema | Invertogram / distal colostogram |
| Gold standard Dx | Clinical + X-ray | Clinical + X-ray | Rectal biopsy (absent ganglion cells) | Clinical + perineal inspection |
| Emergency complication | Malrotation/volvulus | Short gut (type IIIb) | Enterocolitis (shock, death) | Associated urinary obstruction |
| Surgery | Duodenoduodenostomy | Resection + anastomosis | Pull-through (Swenson/Soave/Duhamel) | PSARP (Peña) ± prior colostomy |
| Key association | Down syndrome | Cystic fibrosis | Down syndrome, RET gene | VACTERL |
| Prognosis | >93% survival | >93% survival | Good with early repair | Depends on level; low = good continence |
PART 6: EXAM-FOCUSED DISCUSSION QUESTIONS
- A neonate presents with bilious vomiting and a "double bubble" on X-ray. What is the diagnosis, and what associated condition must you screen for immediately?
- Explain why the DILATED bowel in Hirschsprung's disease is actually the NORMAL bowel.
- What is the gold standard test for Hirschsprung's, and what are the histological findings?
- A female neonate is found to have a single perineal orifice for urine and stool. What is the diagnosis and how do you manage it?
- Differentiate the pathogenesis of duodenal atresia from jejunoileal atresia.
- What is HAEC? Why is it the most dangerous complication of Hirschsprung's?
- Name the three classic pull-through procedures for Hirschsprung's and briefly describe each.
- What does the VACTERL association stand for and when is it relevant?
- How does the Grosfeld classification help guide surgery in jejunoileal atresia?
- A male neonate has no visible anal opening. Meconium is seen in the urine. What type of defect does this indicate, and what is your management plan?
REFERENCES
- Sabiston Textbook of Surgery, 21st Edition — Chapter 117: Pediatric Surgery (pp. 2682–2690) — primary source for all content above
- Schwartz's Principles of Surgery, 11th Edition — Pediatric intestinal atresia, Hirschsprung's disease
- Robbins & Cotran Pathologic Basis of Disease — Hirschsprung's pathology, aganglionic megacolon
- Bailey & Love's Short Practice of Surgery, 28th Edition — Congenital anomalies
Write prescription for captopril tabletb 25 mg according to kazakstan
РЕЦЕПТ / PRESCRIPTION
Қазақстан Республикасы / Republic of Kazakhstan
| Күні / Date: | ___ / ___ / 20___ |
| Науқастың аты-жөні / Patient's Full Name: | _________________________________ |
| Жасы / Age: | _______ жыл / years |
| Мекенжайы / Address: | _________________________________ |
Rp.:
Captopril 0.025 (25 mg) D.t.d. № 30 tab. S. По ½ таблетки (12,5 мг) 2–3 раза в день за 30–60 минут до еды, под контроль АД. (½ tablet 2–3 times daily, 30–60 min before meals, under BP monitoring)
📋 Notes on Kazakhstan Prescription Standards:
| Element | Requirement |
|---|---|
| Form used | Form № 107-1/у — standard outpatient prescription form (for non-narcotic medications) |
| Language | Kazakh or Russian (bilingual accepted) |
| Drug name | Written in Latin (Rp.) |
| Dose written in grams | 25 mg = 0.025 in Latin prescription format |
| D.t.d. | Detur tales doses — give such doses (quantity dispensed) |
| S. (Signa) | Instructions for the patient in Russian/Kazakh |
| Validity | Standard prescription valid for 2 months from date of issue |
| Stamp | Institutional round stamp required |
| Signature | Doctor's personal signature mandatory |
💊 Captopril 25 mg — Key Clinical Notes:
| Parameter | Detail |
|---|---|
| Drug class | ACE inhibitor |
| Indications | Hypertension, heart failure, diabetic nephropathy, post-MI left ventricular dysfunction |
| Typical adult dose | 12.5–25 mg 2–3× daily; up to 50 mg 3× daily |
| Pediatric dose | 0.3–0.5 mg/kg/dose, 1–3× daily (titrate carefully) |
| Administration | 30–60 minutes before meals (food reduces absorption by 30–40%) |
| Contraindications | Pregnancy, bilateral renal artery stenosis, history of ACE inhibitor-induced angioedema, hyperkalemia |
| Key side effects | Dry cough, hyperkalemia, first-dose hypotension, angioedema |
| Monitoring | BP, renal function (creatinine, eGFR), serum K⁺ |
Write me prespricption accordingly to pharamcology i have to show it to teacher
РЕЦЕПТ
Full Pharmacology Format Breakdown:
Rp.: (Recipe — "Take")
Rp.: Captoproli 0,025
D.t.d. № 30 in tabulettis
S.: По 1 таблетке 2–3 раза в день
за 30–60 минут до еды,
под контролем артериального давления.
📖 Each Line Explained (for your teacher):
| Latin Abbreviation | Full Latin | Meaning |
|---|---|---|
| Rp.: | Recipe | "Take" — begins every prescription |
| Captoproli | — | Genitive case of drug name in Latin |
| 0,025 | — | Dose in grams (25 mg = 0,025 g) — always in grams in pharmacology |
| D.t.d. № 30 | Detur tales doses | "Give such doses, quantity 30" |
| in tabulettis | — | "In tablets" — dosage form in Latin ablative |
| S.: | Signa | "Label / Instructions for patient" |
✅ Pharmacology Rules Applied:
| Rule | Applied |
|---|---|
| Drug name in Latin genitive case | Captoproli ✓ |
| Dose in grams (not mg) | 0,025 ✓ |
| D.t.d. with quantity | № 30 ✓ |
| Dosage form in Latin ablative | in tabulettis ✓ |
| S. — patient instruction in Russian | ✓ |
| Doctor signature + stamp | ✓ |
| Date written | ✓ |
💊 Pharmacological Justification (for oral defense):
| Parameter | Detail |
|---|---|
| Group | ACE inhibitor (Angiotensin-Converting Enzyme Inhibitor) |
| Mechanism | Blocks conversion of Angiotensin I → Angiotensin II; reduces vasoconstriction and aldosterone secretion → ↓ BP, ↓ preload & afterload |
| Indication | Arterial hypertension, CHF, diabetic nephropathy, post-MI cardioprotection |
| Dose | 25 mg × 2–3 times/day; start with 12.5 mg if elderly or HF |
| Route | Per os (oral), 30–60 min before meals |
| Side effects | Dry cough (bradykinin accumulation), hyperkalemia, first-dose hypotension, angioedema |
| Contraindications | Pregnancy (teratogenic), bilateral renal artery stenosis, angioedema history |
| Drug interactions | + K-sparing diuretics → hyperkalemia; + NSAIDs → reduced efficacy |
In English please
PRESCRIPTION
Rp.: Captoproli 0.025
D.t.d. № 30 in tabulettis
S.: 1 tablet 2–3 times daily,
30–60 minutes before meals,
under blood pressure monitoring.
📖 Each Line Explained:
| Abbreviation | Full Latin Term | Meaning |
|---|---|---|
| Rp.: | Recipe | "Take" — opens every prescription |
| Captoproli | — | Drug name in Latin genitive case |
| 0.025 | — | Dose in grams (25 mg = 0.025 g) |
| D.t.d. № 30 | Detur tales doses | "Give such doses, 30 tablets total" |
| in tabulettis | — | "In tablets" — dosage form in Latin |
| S.: | Signa | "Instructions for the patient" |
✅ Pharmacology Rules Applied:
| Rule | Status |
|---|---|
| Drug name in Latin genitive case | Captoproli ✓ |
| Dose written in grams not mg | 0.025 ✓ |
| D.t.d. with number of tablets | № 30 ✓ |
| Dosage form in Latin ablative | in tabulettis ✓ |
| S. — clear patient instruction | ✓ |
| Doctor signature + institutional stamp | ✓ |
💊 Pharmacological Justification (for oral defense):
| Parameter | Detail |
|---|---|
| Drug class | ACE Inhibitor (Angiotensin-Converting Enzyme Inhibitor) |
| Mechanism of action | Blocks the enzyme that converts Angiotensin I → Angiotensin II → reduces vasoconstriction, reduces aldosterone secretion → lowers blood pressure, reduces preload and afterload on the heart |
| Indications | Arterial hypertension, Chronic heart failure, Diabetic nephropathy, Post-MI cardioprotection |
| Dose | 25 mg × 2–3 times per day; start with 12.5 mg in elderly or heart failure patients |
| Route of administration | Per os (oral), 30–60 minutes before meals (food reduces absorption by ~30%) |
| Side effects | Dry cough (due to bradykinin accumulation), hyperkalemia, first-dose hypotension, angioedema (rare but dangerous) |
| Contraindications | Pregnancy (causes fetal harm), bilateral renal artery stenosis, history of ACE inhibitor-induced angioedema, hyperkalemia |
| Drug interactions | With K-sparing diuretics → risk of hyperkalemia; with NSAIDs → reduced antihypertensive effect; with diuretics → risk of severe first-dose hypotension |
✍️ Clean Copy for Your Notebook:
Date: ___/___/20___
Patient: _______________________ Age: ___
Rp.: Captoproli 0.025
D.t.d. № 30 in tabulettis
S.: 1 tablet 2–3 times daily,
30–60 minutes before meals,
under blood pressure control.
Doctor: _______________________
Signature: ____________________
Stamp: [ ]
Try again