Inclusion body myositis
Reading File
search_medical_text
inclusion body myositis clinical features diagnosis pathophysiology
search_medical_text
inclusion body myositis treatment management prognosis
search_medical_image
inclusion body myositis muscle biopsy histology
GradeResponse
GradeResponse
GradeResponse
Inclusion Body Myositis (IBM)
Overview
Inclusion body myositis is the most common acquired myopathy in adults over 50, and is distinct from other inflammatory myopathies (polymyositis, dermatomyositis) in its clinical pattern, resistance to immunotherapy, and unique pathological features. It has both degenerative and inflammatory components.
Epidemiology
- Prevalence: ~5–70 per million (most common inflammatory myopathy in those >50 years)
- Male predominance (~3:1 male to female ratio)
- Onset typically after age 50; rarely before age 45
- Slowly progressive; most patients require ambulatory aids within 10–15 years
Pathogenesis
IBM has a dual pathology — both inflammatory and degenerative — which is why it does not respond to immunosuppression the way polymyositis does.
Inflammatory component:
- CD8+ cytotoxic T cells invade MHC-I expressing, non-necrotic muscle fibers (same as polymyositis)
- Clonally expanded, antigen-driven T-cell response
Degenerative component:
- Abnormal protein aggregation within muscle fibers: amyloid precursor protein (APP), phosphorylated tau, TDP-43, p62, ubiquitin
- Formation of rimmed vacuoles and tubulofilamentous inclusions (15–21 nm diameter on electron microscopy)
- Cytochrome oxidase (COX)-negative fibers reflect mitochondrial dysfunction
- Resembles a muscle "proteinopathy" akin to neurodegenerative disease
Clinical Features
IBM has a characteristic pattern that distinguishes it from other myopathies (Harrison's, p. 10284 / index p. 14547):
| Feature | Description |
|---|---|
| Finger flexor weakness | Early, prominent — difficulty gripping, opening jars, turning keys |
| Quadriceps weakness | Early — frequent falls, difficulty rising from chairs |
| Dysphagia | Up to 40–80% — from pharyngeal and esophageal involvement |
| Distal + proximal weakness | Asymmetric; distal involvement unusual for other myopathies |
| Foot drop | Due to tibialis anterior weakness |
| Facial weakness | Occurs in some patients |
| Atrophy | Prominent forearm and quadriceps wasting |
Key distinguishing point: asymmetric, distal > proximal weakness with selective finger flexor + quadriceps involvement is virtually pathognomonic for IBM.
Diagnosis
Diagnostic Criteria (2011 ENMC Criteria — key elements)
- Age >45, duration >12 months
- Finger flexor or quadriceps weakness
- Biopsy showing endomysial inflammation + rimmed vacuoles OR p62/TDP-43 inclusions
Laboratory Findings
- CK: Normal or mildly elevated (up to 10× normal; often <1000 U/L — lower than polymyositis)
- Anti-cN-1A antibody (anti-NT5C1A): Present in ~33–70%; not diagnostic alone but supports IBM
- ANA and myositis-specific antibodies (anti-Jo-1, etc.) typically negative
EMG
- Mixed pattern: myopathic + neuropathic features (short-duration and long-duration MUAPs, fibrillations)
MRI
- Selective involvement of quadriceps (especially rectus femoris spared relative to vastus lateralis) and forearm flexors
Muscle Biopsy — Gold Standard
Findings (Harrison's, p. 10284):
- Rimmed vacuoles lined by granular material (H&E)
- Endomysial inflammation (CD8+ T cells invading non-necrotic fibers)
- COX-negative fibers (mitochondrial dysfunction)
- p62-positive cytoplasmic inclusions
- Tubulofilamentous inclusions (15–21 nm) on electron microscopy
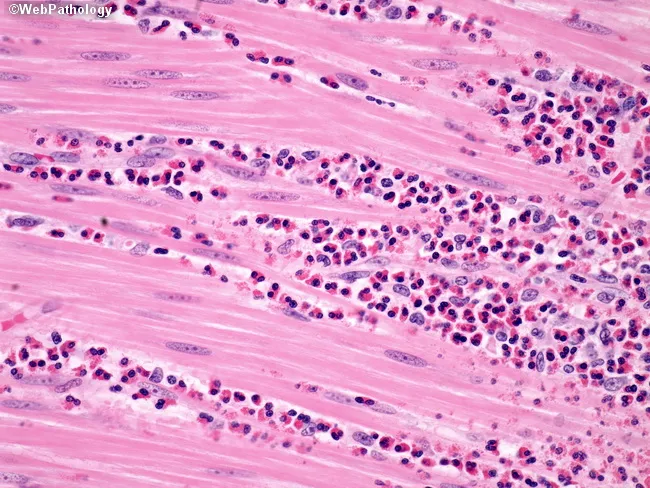
H&E biopsy showing endomysial lymphocytic infiltration and myofiber degeneration, characteristic of inflammatory myopathy including IBM.
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| Polymyositis | No rimmed vacuoles; responds to steroids; no distal predominance |
| Dermatomyositis | Skin findings (heliotrope rash, Gottron's papules); perifascicular atrophy on biopsy |
| LOPD (late-onset Pompe) | Acid maltase deficiency; vacuolar myopathy; no inflammation; enzyme assay diagnostic |
| IBMPFD | Paget's disease + frontotemporal dementia + myopathy; VCP gene mutation |
| ALS/motor neuron disease | UMN + LMN signs; no biopsy inflammation |
| Oculopharyngeal MD | PABPN1 mutation; ptosis + dysphagia; smaller rimmed vacuoles |
Management
Pharmacological
- No proven disease-modifying therapy — IBM is largely refractory to immunosuppression
- Corticosteroids: Trials consistently show no sustained benefit; may transiently improve inflammation but not function
- IVIG: May modestly benefit dysphagia in some patients; no effect on limb strength
- Other immunosuppressants (methotrexate, azathioprine, cyclosporine): No proven benefit
- Bimagrumab (anti-ActRII antibody): Showed increased lean mass in trials; ongoing investigation
- Rapamycin (sirolimus): Phase 2 trials exploring the autophagy/mTOR pathway — early results modestly promising
Non-Pharmacological (cornerstone of care)
- Physical therapy: Resistance training and aerobic exercise are safe and may slow functional decline
- Occupational therapy: Adaptive devices for hand function (jar openers, key turners)
- Speech therapy + swallowing evaluation: Critical for dysphagia; PEG tube if severe
- Orthotics: Ankle-foot orthoses (AFOs) for foot drop
- Fall prevention: Home modifications, walking aids
Prognosis
- Slowly but relentlessly progressive — no spontaneous remission
- Most patients lose independent ambulation within 10–15 years of symptom onset
- Dysphagia is a major source of morbidity (aspiration pneumonia)
- Respiratory muscle involvement occurs in advanced disease
- Life expectancy modestly reduced; leading causes of death include aspiration pneumonia and falls
Key Distinguishing Points vs. Other IIMs
| Feature | IBM | Polymyositis | Dermatomyositis |
|---|---|---|---|
| Age of onset | >50 | Any age | Any age |
| Sex | M > F | F > M | F > M |
| Weakness pattern | Distal + proximal, asymmetric | Proximal, symmetric | Proximal, symmetric |
| CK | Normal–mild ↑ | High | High |
| Rimmed vacuoles | Yes | No | No |
| Response to steroids | No | Yes | Yes |
| Anti-cN-1A antibody | Yes (33–70%) | No | No |
Sources: Harrison's Principles of Internal Medicine, 21st Ed., pp. 10284, 14547; supplemented from current IBM clinical literature.
This is a shared conversation. Sign in to Orris to start your own chat.