Visual study aid for sickle cell disease pathology
Reading File
search_medical_text
sickle cell disease pathophysiology molecular basis vaso-occlusion hemolysis
search_medical_image
sickle cell disease pathology blood smear sickling vaso-occlusion
GradeResponse
GradeResponse
Here is a comprehensive visual study aid for sickle cell disease (SCD) pathology.
Sickle Cell Disease — Pathology Study Aid
1. The Molecular Root Cause
| Feature | Detail |
|---|---|
| Mutation | Single point mutation: GAG → GTG in codon 6 of the β-globin gene (chromosome 11) |
| Amino acid change | Glutamate → Valine at position 6 of the β-globin chain |
| Abnormal Hb | HbS (α₂β₂^S) instead of normal HbA (α₂β₂) |
| Inheritance | Autosomal recessive; HbSS = disease, HbAS = trait (carrier) |
Key insight: The valine substitution creates a hydrophobic "sticky patch" on the β-chain surface. When deoxygenated, this patch allows HbS molecules to polymerize into long rigid fibers.
2. The Central Cascade: Deoxygenation → Polymerization → Sickling
Low pO₂ (e.g., capillaries, exertion, infection)
↓
HbS deoxygenated → hydrophobic Val-6 exposed
↓
HbS molecules polymerize into long fiber bundles
↓
Erythrocyte deforms → SICKLE shape
↓
┌──────────────┴──────────────┐
│ │
Re-oxygenated Repeated sickling cycles
(reversible sickling) │
↓
Irreversibly Sickled Cells (ISCs)
(membrane permanently damaged)
Concentration matters: Polymerization depends on the 30th power of Hb concentration. Even tiny increases in cell dehydration dramatically accelerate sickling (Harrison's, p. 2915).
3. Blood Smear — What You See
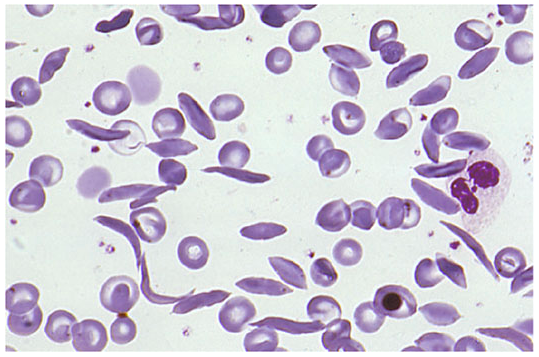
Wright-Giemsa stained peripheral blood smear in SCD. Key findings:
| Finding | Significance |
|---|---|
| Sickle/crescent-shaped RBCs | HbS polymerization under deoxygenation |
| Irreversibly sickled cells (ISCs) | Permanently deformed; membrane-damaged even when re-oxygenated |
| Target cells | Thin, deformable cells; also seen in HbC disease and thalassemia |
| Polychromasia | Reticulocytosis — compensatory response to hemolysis |
| Howell-Jolly bodies | Functional asplenia (autosplenectomy) after repeated infarcts |
| Nucleated RBCs | Seen during hemolytic crises; bone marrow stress response |
4. Two Major Pathophysiological Arms
ARM 1: Vaso-occlusion
Sickled RBCs
↓ rigid, adhesive
Stick to vascular endothelium
↓
Occlude capillaries and postcapillary venules
↓
Ischemia → Infarction → End-organ damage
- Sickled cells express abnormal adhesion molecules (e.g., αVβ3 integrin, BCAM/LU)
- Leukocytes and platelets are also activated, amplifying occlusion
- Stasis worsens local deoxygenation → more sickling (vicious cycle)
ARM 2: Hemolytic Anemia
Sickled RBCs
↓ membrane damage, oxidative stress
Shortened RBC lifespan: ~10–20 days (vs. normal 120 days)
↓
Intravascular + extravascular hemolysis
↓
↓ Hemoglobin in plasma → Scavenges NO
↓
Endothelial dysfunction, vasospasm, pulmonary hypertension
5. Complications Map by Organ System
| System | Complication | Mechanism |
|---|---|---|
| Blood | Chronic hemolytic anemia (Hb ~6–9 g/dL) | RBC destruction |
| Spleen | Autosplenectomy (by ~5 yrs) | Repeated infarcts → fibrosis |
| Bone | Vaso-occlusive pain crisis; avascular necrosis (femoral head) | Marrow/cortical infarction |
| Bone marrow | Aplastic crisis (Parvovirus B19) | Suppression of erythropoiesis |
| Brain | Stroke (ischemic in children; hemorrhagic in adults) | Large & small vessel occlusion |
| Lung | Acute Chest Syndrome (ACS) | Fat embolism + infection + infarction |
| Kidney | Papillary necrosis, hematuria, CKD, hyposthenuria | Medullary hypoxia/infarction |
| Eye | Proliferative retinopathy ("sea fan" neovascularization) | Retinal ischemia |
| Penis | Priapism | Venous stasis in corpora cavernosa |
| Liver/GB | Pigment gallstones (bilirubin) | Chronic hemolysis → ↑ unconjugated bilirubin |
| Heart | Cardiomegaly, high-output failure | Chronic anemia |
6. Triggers That Precipitate Crises
| Trigger | Why It Causes Sickling |
|---|---|
| Infection / fever | ↑ O₂ demand, dehydration |
| Dehydration | ↑ intracellular HbS concentration → faster polymerization |
| Hypoxia (altitude, surgery, sleep apnea) | Direct deoxygenation of HbS |
| Cold / temperature change | Vasoconstriction → stasis |
| Acidosis | Bohr effect → rightward O₂ dissociation curve → more deoxyHbS |
| Stress / exertion | Local tissue hypoxia |
7. Lab Findings Summary
| Test | Finding | Why |
|---|---|---|
| CBC | Hb 6–9 g/dL; ↑ reticulocytes (10–25%) | Chronic hemolysis + compensatory erythropoiesis |
| Peripheral smear | Sickle cells, target cells, Howell-Jolly bodies | (see above) |
| Bilirubin | ↑ indirect (unconjugated) | RBC breakdown |
| LDH | Elevated | Intravascular hemolysis |
| Haptoglobin | Low/absent | Binds free Hb; depleted in hemolysis |
| Hemoglobin electrophoresis | HbS >90%, absent HbA; HbF variable | Definitive diagnosis |
| Urinalysis | Isosthenuria, hematuria | Renal medullary damage |
8. HbF — The Protective Factor
- Fetal hemoglobin (HbF, α₂γ₂) does not polymerize with HbS
- Higher HbF levels = less sickling = milder disease
- Hydroxyurea (first-line therapy) works by reactivating γ-globin gene expression → ↑ HbF
- This is why some patients with HbSS + high HbF have surprisingly mild phenotypes
9. Quick Comparison: SCD Genotypes
| Genotype | Disease Severity | HbS% | Notes |
|---|---|---|---|
| HbSS | Most severe | ~90–95% | Classic sickle cell anemia |
| HbSC | Moderate | ~50% HbS, ~50% HbC | More retinopathy, less anemia |
| HbS/β⁰-thal | Severe (similar to HbSS) | High HbS, absent HbA | No normal β-globin produced |
| HbS/β⁺-thal | Mild–moderate | HbS + some HbA | Some normal β-globin produced |
| HbAS | Carrier (trait) | ~40% HbS | Usually asymptomatic; protective vs. P. falciparum |
Sources: Harrison's Principles of Internal Medicine, 21st Edition (pp. 2915, 1726)
How to generate poster
To create a visual poster from this study aid, you have several options depending on your tools:
Option 1: Canva (Easiest, No Code)
- Go to canva.com → Create design → Poster
- Choose a medical/infographic template
- Paste each section (molecular cause, cascade, complications table, etc.) into text boxes
- Add the blood smear image by URL:
https://cdn.orris.care/cdss_images/HARRISON_1763036071170_b52af39c-ae90-4f8b-9826-2f200ccfcfd6.png - Export as PDF or PNG
Option 2: Python (Automated, Programmatic)
Using matplotlib or reportlab to generate a PDF poster programmatically. I can write this code for you — it would:
- Lay out all sections as styled panels
- Embed the blood smear image
- Export a print-ready PDF
Option 3: HTML/CSS → Print to PDF
I can generate a single self-contained HTML file you open in a browser and print (Ctrl+P → Save as PDF). This gives full styling control with color-coded sections, tables, and images.
Option 4: PowerPoint / Google Slides
- Set slide size to A1 or A0 (poster dimensions) under Page Setup
- Paste sections into grouped text boxes
- Use a dark navy or white background with color-coded panels per system
Which would you prefer?
- A) Python script (generates a PDF automatically)
- B) HTML file (open in browser, print to PDF)
- C) Canva layout instructions (step-by-step)
- D) PowerPoint template structure
This is a shared conversation. Sign in to Orris to start your own chat.