Now I have excellent retrieved content plus comprehensive knowledge to build a thorough, well-illustrated class resource.
Malformations of the Musculoskeletal System in Children
Complete Class Discussion Notes
1. DEFINITION
Congenital musculoskeletal malformations are structural abnormalities of bones, joints, cartilage, muscles, ligaments, or connective tissue that arise during embryonic/fetal development and are present at birth or manifest during childhood growth.
They are broadly categorized using the Spranger Classification:
| Category | Mechanism | Examples |
|---|
| Malformation | Intrinsic error in morphogenesis | Clubfoot, DDH, polydactyly |
| Deformation | Extrinsic mechanical forces on structurally normal tissue | Positional foot deformity |
| Disruption | Breakdown of originally normal tissue | Amniotic band syndrome, limb reduction |
| Dysplasia | Abnormal organization of cells into tissue | Achondroplasia, OI |
2. EMBRYOLOGICAL BASIS
Limb development occurs between weeks 4–8 of gestation. Three key organizers govern normal development:
| Organizer | Location | Controls |
|---|
| Apical Ectodermal Ridge (AER) | Distal limb tip | Proximodistal outgrowth |
| Zone of Polarizing Activity (ZPA) | Posterior mesenchyme | Anteroposterior patterning (digit identity) |
| Dorsoventral Ectoderm | Dorsal/ventral surface | Dorsoventral patterning |
Disruption of any of these axes during this window produces the corresponding malformation. The earlier the insult, the more severe and complex the result.
3. GENERAL ETIOLOGY
| Category | Mechanism | Examples |
|---|
| Genetic / Single gene | Mutation in developmental gene | FGFR3 → achondroplasia; COL1A → OI; SHH pathway → polydactyly |
| Chromosomal | Trisomies, deletions | Trisomy 18, 21 → multiple anomalies |
| Multifactorial | Gene + environment threshold | DDH, clubfoot, idiopathic scoliosis |
| Teratogenic | Drug, chemical, infection | Valproic acid, thalidomide, rubella |
| Mechanical / Intrauterine | Oligohydramnios, abnormal lie, twinning | Positional deformities, DDH |
| Vascular disruption | Ischemia in early gestation | Transverse limb defects |
4. MAJOR CONDITIONS
A. Developmental Dysplasia of the Hip (DDH)
Definition
A spectrum of hip abnormalities ranging from mild acetabular dysplasia → subluxation → complete dislocation, due to inadequate development of the hip socket and/or femoral head.
Etiology & Risk Factors
| Risk Factor | Relative Risk |
|---|
| Female sex | 4–8× |
| Breech presentation | 10–20× |
| Family history (1st-degree) | 12× |
| First-born | Tight, unprepared uterus |
| Left hip (most common 60%) | Lies against maternal spine in LOA position |
| Oligohydramnios | Mechanical constraint |
| Ligamentous laxity (maternal estrogen) | Neonatal hip instability |
Pathogenesis
Shallow/underdeveloped acetabulum
↓
Femoral head inadequately contained
↓
Head migrates superolaterally
↓
Secondary changes:
• Pulvinar (fibrofatty tissue) fills acetabulum
• Labrum everts/inverts (limbus)
• Capsule elongates and hourglass constricts
• Iliopsoas & adductors shorten
↓
If untreated → secondary OA by 3rd–4th decade
Clinical Features
- Neonate: Asymmetric skin folds, apparent leg shortening (Galeazzi sign), limited hip abduction
- Ortolani test: Hip reduced → gentle abduction → dislocated head clunks back in ("in sign")
- Barlow test: Hip reduced → adduction + posterior pressure → head clunks out ("out sign")
- Walking child: Trendelenburg gait, waddling (bilateral), apparent leg shortening
- Note: Ortolani/Barlow become negative after 6–8 weeks as soft tissues tighten — do not exclude DDH
Diagnosis
| Age | Investigation | Rationale |
|---|
| 0–4 months | Ultrasound (Graf method) | Ossific nucleus absent; X-ray useless |
| 4–6 months | Ultrasound transitioning to X-ray | |
| > 6 months | AP pelvis X-ray | Ossific nucleus visible |
Graf Ultrasound Classification:
- Type I: Normal (α angle > 60°)
- Type IIa: Immature (α 50–59°, age < 3 months — physiological)
- Type IIb: Delayed ossification (α 50–59°, age > 3 months — pathological)
- Type III: Subluxed cartilaginous roof
- Type IV: Dislocated
X-ray Lines (for > 6 months):
- Hilgenreiner line: Horizontal through both triradiate cartilages
- Perkin line: Vertical from lateral acetabular edge
- Normal: ossific nucleus in inner lower quadrant
- Shenton's line: Disrupted in subluxation/dislocation
Treatment
| Age | Treatment | Notes |
|---|
| 0–6 months | Pavlik harness | Maintains flexion 90–100°, abduction 40–50°; success ~95% in neonates; check for femoral nerve palsy |
| 6–18 months | Closed reduction + hip spica cast under GA | Arthrogram to confirm reduction; cast 12 weeks |
| 18 months – 3 yrs | Open reduction ± femoral shortening | Approach: medial (Ludloff) or anterior (Smith-Petersen) |
| > 3 yrs | Open reduction + femoral + pelvic osteotomy | Salter (< 6 yrs), Pemberton, Dega, Triple osteotomy |
Complications of treatment: Avascular necrosis (AVN) of femoral head — most feared complication; risk ↑ with forced/over-abduction
B. Congenital Talipes Equinovarus (CTEV / Clubfoot)
Definition
A rigid, complex three-dimensional foot deformity present at birth. Components remembered as CAVE:
- C — Cavus (high arch of midfoot)
- A — Adductus (forefoot adducted)
- V — Varus (hindfoot inverted)
- E — Equinus (ankle plantarflexed)
Epidemiology
- 1–2 per 1,000 live births; male 2:1; bilateral in ~50%
- Most common congenital foot deformity
Etiology
| Type | Details |
|---|
| Idiopathic (80%) | Multifactorial; 2–4% sibling recurrence; 10× if parent + sibling affected |
| Neurogenic | Spina bifida (myelomeningocele), cerebral palsy — rigid, worse prognosis |
| Syndromic | Arthrogryposis (fibrous joint ankylosis), Larsen syndrome, chromosomal anomalies |
| Positional | In utero compression — flexible, corrects with gentle manipulation |
Pathogenesis
- Primary defect: deviated talar neck (medially and plantarward rotated)
- All other bones displace around the abnormal talus:
- Calcaneus: inverted, equinus position
- Navicular: displaced medially onto medial talar neck
- Cuboid: follows calcaneus medially
- Contracted structures: tibialis posterior, FHL, FDL, plantar fascia, spring ligament, deltoid ligament, posterior capsule
- Relatively weak/lengthened: peronei, anterior tibialis, extensor tendons
Assessment
Pirani Scoring System (0–6 scale, guides Ponseti treatment):
- Midfoot score (3 points): Curved lateral border, medial crease, talar head coverage
- Hindfoot score (3 points): Posterior crease, rigid equinus, empty heel
Dimeglio Classification (I–IV): Based on reducibility
- Grade I: Soft/postural (benign)
- Grade II: Soft/resistant (moderate)
- Grade III: Resistant/soft (severe)
- Grade IV: Rigid (very severe)
X-ray: Talocalcaneal angle (Kite's angle) < 20° (normal 20–40°); talus-first metatarsal angle (Meary's) abnormal
Treatment
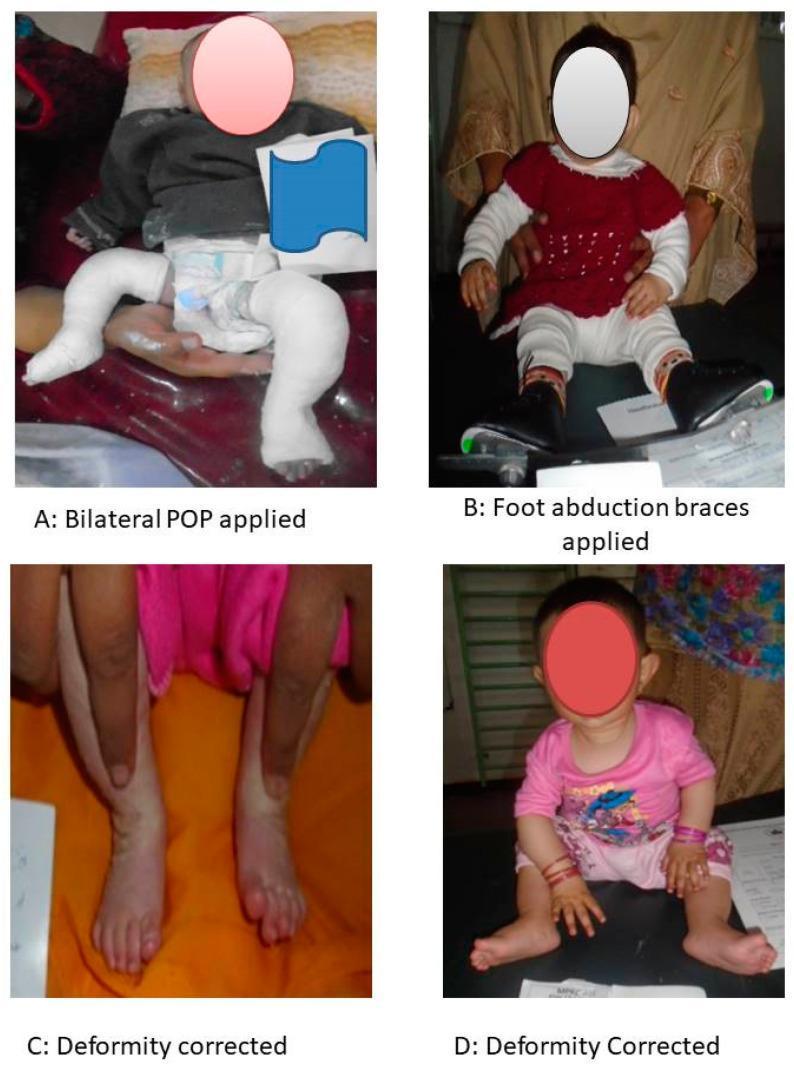
Ponseti Method (Gold Standard — > 95% success):
Ponseti method: (A) Serial POP long-leg casts correcting CAVE sequentially. (B) Denis Browne foot abduction brace for maintenance. (C–D) Corrected feet showing plantigrade positioning with no residual adduction. (PMC Clinical VQA)
- Serial casting (weekly, 5–7 casts): Corrects C → A → V in sequence; hindfoot corrects last
- Percutaneous Achilles tenotomy (in ~85–90%): Corrects equinus; local anesthesia; cast for 3 weeks post-procedure
- Foot Abduction Brace (Denis Browne orthosis): 70° abduction, 10° dorsiflexion; 23 hrs/day × 3 months, then nights/naps until age 4–5 years
- Non-compliance = #1 cause of relapse
Surgical (relapse/resistant cases):
- Tibialis anterior tendon transfer to lateral cuneiform — corrects dynamic supination
- Posteromedial soft-tissue release (McKay/Carroll) — historically used; now reserved for failures
C. Congenital Scoliosis
Definition
Lateral spinal curvature caused by structural vertebral anomalies present at birth — distinct from idiopathic scoliosis which has normal vertebrae.
Etiology
- Failure of normal somite differentiation at weeks 5–8 of gestation
- Environmental triggers: maternal hyperthermia, diabetes, antiepileptic drugs
- Associated with VACTERL syndrome (Vertebral, Anorectal, Cardiac, Tracheo-Esophageal, Renal, Limb)
- Spinal cord anomalies in 18–38% (diastematomyelia, tethered cord, syringomyelia) — always get MRI
Classification (Winter)
| Type | Defect | Examples | Prognosis |
|---|
| Type I | Failure of formation | Hemivertebra (fully/partially segmented) | Variable — depends on hemivertebra type |
| Type II | Failure of segmentation | Unilateral unsegmented bar (block vertebra on one side) | Worst — relentlessly progressive; tethers convex side |
| Type III | Mixed | Bar + hemivertebra on opposite side | Most severe progression |
Hemivertebra subtypes:
- Fully segmented (free growth plates both ends) — most progressive
- Semi-segmented — moderate progression
- Incarcerated (wedged between normal vertebrae) — minimal progression
- Non-segmented (fused) — least progressive
Pathogenesis
Vertebral anomaly (e.g. unsegmented bar)
↓
Asymmetric longitudinal growth
↓
Progressive lateral curve + rotation
↓
Rib cage deformity → Thoracic Insufficiency Syndrome
↓
Restrictive lung disease + cor pulmonale (severe cases)
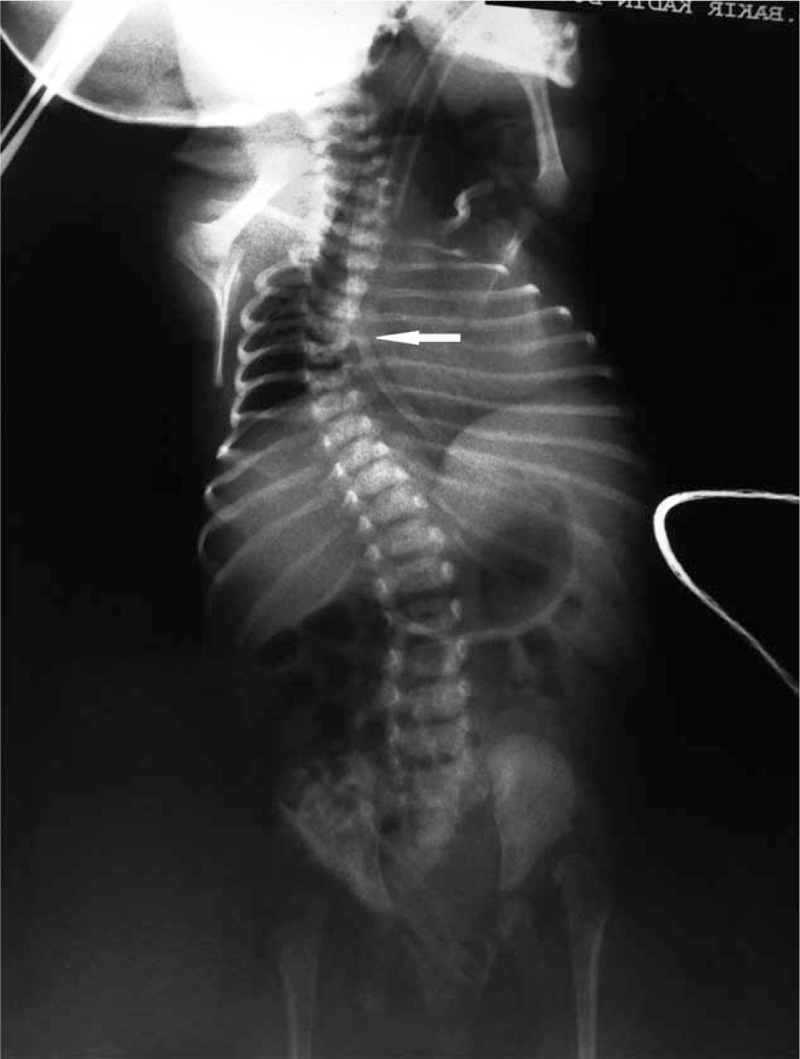
The radiograph below demonstrates congenital scoliosis with a unilateral unsegmented bar causing a right-sided thoracic curve, narrowed thoracic cage, and bilateral hip dislocations — illustrating multi-system skeletal dysplasia:
AP radiograph: Right thoracic congenital scoliosis with unilateral unsegmented bar (white arrow), bilateral developmental dislocation of hips, and reduced thoracic volume — indicating thoracic insufficiency syndrome. (ROCO Radiology)
Diagnosis
- Adams Forward Bend Test: rib hump/trunk asymmetry
- Standing AP + lateral X-ray: Cobb angle; vertebral anomaly characterization
- MRI whole spine: Mandatory before any surgery — intraspinal anomalies
- CT 3D reconstruction: Surgical planning
- Echocardiogram + renal ultrasound: VACTERL screening
- Pulmonary function tests (older children)
Treatment
| Cobb Angle / Age | Treatment |
|---|
| < 20°, low risk | Observation (6-monthly X-rays) |
| Progressive, young child | Growing rods (magnetically controlled — MAGEC rods) distracted every 6 months; preserves growth |
| Thoracic insufficiency syndrome | VEPTR (Vertical Expandable Prosthetic Titanium Rib) |
| Fully segmented hemivertebra | Hemivertebra resection (< 5 years, before curve is established) |
| Near skeletal maturity | Spinal fusion with instrumentation |
D. Osteogenesis Imperfecta (OI) — "Brittle Bone Disease"
Definition
A heritable connective tissue disorder caused by qualitative or quantitative defects in Type I collagen, resulting in abnormal bone fragility, multiple fractures, and progressive skeletal deformity. (Bailey & Love, p. 636)
Pathogenesis
- Mutations in COL1A1 or COL1A2 genes (most common — autosomal dominant)
- Newer forms: CRTAP, LEPRE1, PPIB mutations (autosomal recessive — severe)
- Result: disordered collagen fibril assembly → abnormal bone matrix → osteoporosis + fracture susceptibility
- Bone heals promptly but in disorganized fashion → progressive bowing deformities
- ALL collagen-containing structures affected: bone, ligaments, sclerae, dentin, middle ear ossicles
Sillence Classification
| Type | Severity | Sclerae | Teeth | Features |
|---|
| I | Mild | Blue | Normal/DI | Most common; near-normal stature; fractures with minor trauma |
| II | Lethal perinatal | Dark blue | — | Multiple in-utero fractures; stillbirth or death within days |
| III | Severe progressive | Variable | Often DI | Triangular face; severe bowing; wheelchair-bound; most severe surviving form |
| IV | Moderate | White/normal | Often DI | Variable deformity; short stature |
(DI = Dentinogenesis imperfecta)
Diagnosis
- Clinical triad: Fractures disproportionate to trauma + blue sclerae + dentinogenesis imperfecta
- X-ray: Generalized osteopenia, wormian bones (skull), bowed long bones, vertebral compression fractures
- DXA scan: Reduced bone mineral density (BMD) Z-score
- Molecular genetic testing: COL1A1/COL1A2 sequencing
- Skin biopsy: Collagen biochemical analysis (historical)
- Critical differential: Non-accidental injury (NAI/child abuse) — history inconsistency, dating of fractures, multidisciplinary team assessment essential
Treatment
| Modality | Details |
|---|
| Bisphosphonates | IV pamidronate or zoledronate — cyclic therapy; increases BMD, reduces fracture frequency and bone pain; standard of care for moderate–severe OI |
| Physiotherapy | Hydrotherapy, muscle strengthening, standing frames — prevent disuse atrophy |
| Intramedullary rodding | Fassier-Duval telescoping rods — correct bowing, stabilize long bones, prevent fractures; rods expand with growth |
| Denosumab | Emerging — RANK-L inhibitor for bisphosphonate-resistant cases |
| Hearing aids | Conductive hearing loss from ossicle involvement |
| Dental care | Crowning, dental hygiene for dentinogenesis imperfecta |
E. Achondroplasia
Definition
The most common skeletal dysplasia; a rhizomelic (proximal > distal limb shortening) form of dwarfism due to defective endochondral ossification.
Etiology & Pathogenesis
- Autosomal dominant; > 80% are de novo mutations (associated with advanced paternal age)
- Gain-of-function mutation in FGFR3 (fibroblast growth factor receptor 3) gene — most commonly Gly380Arg (c.1138G>A, chromosome 4p16.3)
- FGFR3 normally inhibits chondrocyte proliferation in growth plate; mutant receptor is constitutively active
- Result: severely suppressed endochondral ossification → short long bones
- Intramembranous ossification is normal → normal skull vault and clavicles; skull base (endochondral) → small foramen magnum
Clinical Features
- Rhizomelic limb shortening (humerus/femur disproportionately short)
- Macrocephaly, frontal bossing, midface hypoplasia
- Trident hand configuration
- Thoracolumbar kyphosis in infancy → lumbar hyperlordosis with age
- Normal intelligence; near-normal lifespan
- Homozygous achondroplasia = lethal (respiratory failure)
Complications
| Complication | Mechanism | Management |
|---|
| Foramen magnum stenosis | Small endochondral skull base | High cervical MRI; decompression if symptomatic |
| Obstructive sleep apnea | Midface hypoplasia | Sleep study; CPAP; adenotonsillectomy |
| Spinal canal stenosis | Shortened pedicles | Laminectomy in adulthood |
| Recurrent otitis media | Eustachian tube dysfunction | Grommets |
| Genu varum | Fibular overgrowth relative to tibia | Guided growth / osteotomy |
Diagnosis
- Clinical + skeletal survey X-ray: Shortened tubular bones, "champagne glass" pelvis (narrow sacrosciatic notch), tombstone-shaped iliac wings, small foramen magnum, interpediculate distance narrows caudally (opposite of normal)
- Molecular genetics: FGFR3 point mutation
- Prenatal: Ultrasound (femur length < 5th percentile after 22 weeks); amniocentesis for FGFR3
Treatment
- Vosoritide (CNP analogue; FGFR3 inhibitor) — FDA-approved 2021; daily SC injection; improves annualized height velocity in children ≥ 2 years
- Foramen magnum decompression if symptomatic stenosis
- Guided growth / osteotomy for symptomatic genu varum
- Limb lengthening (Ilizarov/LON technique) — controversial; performed in specialist centres
F. Polydactyly & Syndactyly
Polydactyly — Extra Digits
- Preaxial: Radial/tibial side (thumb/great toe duplication) — often associated with genetic syndromes
- Central: Middle digits — rare, usually syndromic
- Postaxial: Ulnar/fibular side (little finger/5th toe) — most common; Type A (fully formed), Type B (rudimentary)
Pathogenesis: Dysregulation of ZPA — overexpression of Sonic Hedgehog (SHH) signaling → supernumerary digit specification
Treatment:
- Type B postaxial: suture ligation at birth or excision
- Bony types: surgical reconstruction at 1–2 years (before school age)
Syndactyly — Fused Digits
- Most common congenital hand anomaly (1 in 2,000–3,000)
- Simple: skin/soft tissue only
- Complex: bony fusion
- Complete: to fingertip; Incomplete: partial
Pathogenesis: Failure of interdigital programmed apoptosis (regulated by BMP signaling) between weeks 6–8
Treatment:
- Surgical separation + skin grafting at 6–18 months
- Priority: digit pairs of unequal length (ring-little, index-middle) — separate earlier to prevent tethering and angular deformity
G. Congenital Limb Reduction Defects
Definition
Partial or complete absence of a limb or limb segment.
Classification
- Transverse: Complete absence distal to a level (amputation-like)
- Longitudinal: Absence along the limb axis
- Radial hemimelia (radial club hand) — associated with Fanconi anemia, TAR syndrome, Holt-Oram syndrome; screen with CBC and bone marrow biopsy
- Fibular hemimelia — most common longitudinal deficiency; associated with femoral and tarsal anomalies
- Tibial hemimelia — rare; associated with polydactyly and split hand/foot
Etiology
- Vascular disruption in first trimester
- Amniotic band syndrome (Streeter dysplasia)
- Teratogenic: thalidomide (phocomelia — absent proximal + present distal limb), misoprostol
- Chromosomal
Treatment
- Radial hemimelia: Centralization of hand over ulna + pollicization (creating thumb from index finger)
- Fibular hemimelia: Syme amputation (well-accepted in infancy) vs. limb lengthening (Ilizarov); decision based on foot function, leg length discrepancy
- Prosthetic fitting: Age-appropriate; upper limb from 6 months (sitting), lower limb from 9–12 months (standing)
H. Larsen Syndrome (Multi-joint Dislocation)
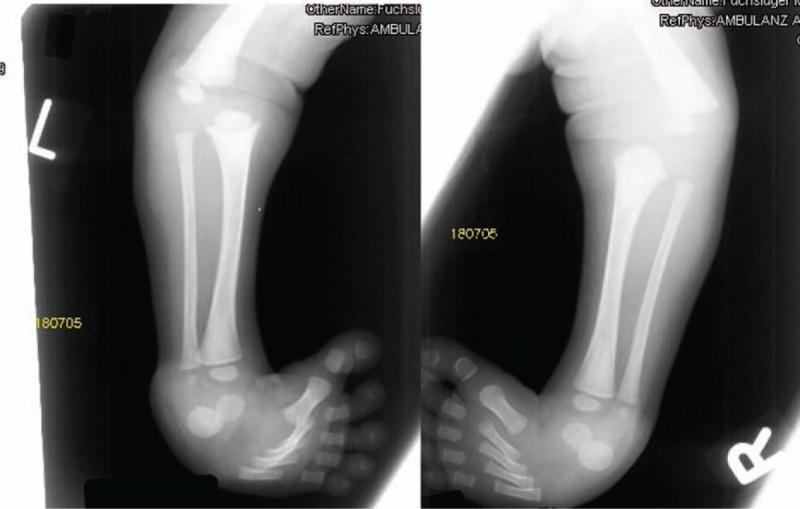
The radiograph below demonstrates this dramatically:
Bilateral anterior knee dislocation (tibia dislocated anteriorly on femur) with bilateral talipes equinovarus. Multiple joint dislocations and ligamentous laxity in skeletal dysplasia/Larsen syndrome. Abnormal ossification center relationships are visible at tarsal bones. (PMC Clinical VQA)
- Mutation in FLNB (filamin B) — regulates cytoskeletal organization
- Multiple large joint dislocations (hips, knees, elbows) + clubfoot + cervical spine instability
- Treatment: serial casting, surgical stabilization, cervical spine fusion if needed
5. UNIFIED DIAGNOSTIC APPROACH
Suspected MSK Malformation
│
├── History: Family history, maternal drugs/illness,
│ birth presentation, fetal movements, amniocentesis results
│
├── Physical Examination:
│ • Dysmorphic features (syndromic?)
│ • Range of motion, flexibility vs rigidity
│ • Neurological exam (spine anomaly?)
│ • Skin: cafe-au-lait, haemangiomas
│
├── Imaging:
│ • X-ray (skeletal survey if dysplasia suspected)
│ • Ultrasound (hips < 4 months; soft tissue/cord)
│ • MRI (spinal cord anomalies, soft tissue detail)
│ • CT 3D (complex bony anomalies, surgical planning)
│
└── Investigations:
• Karyotype / chromosomal microarray
• Gene panel / WES (if syndromic/dysmorphic)
• Echocardiogram + renal USS (VACTERL)
• CBC (radial hemimelia → Fanconi)
6. TREATMENT PRINCIPLES SUMMARY
| Modality | Conditions | Key Points |
|---|
| Serial casting | Clubfoot, congenital knee dislocation | Ponseti; weekly; corrects sequentially |
| Splinting/Bracing | DDH (Pavlik), clubfoot (Denis Browne), scoliosis (TLSO) | Compliance is critical |
| Closed/Open reduction | DDH | Closed first; open if closed fails or > 18 months |
| Osteotomy | DDH (Salter/Pemberton), OI limb bowing | Corrects anatomy; allows normal growth |
| Intramedullary rodding | OI, severe bowing | Fassier-Duval telescoping rods |
| Growing rods / VEPTR | Congenital scoliosis (young children) | Preserves growth; expands every 6 months |
| Spinal fusion | Scoliosis at/near skeletal maturity | Halts progression |
| Tendon transfer | Clubfoot relapse | Tibialis anterior → lateral cuneiform |
| Medical therapy | OI (bisphosphonates), Achondroplasia (vosoritide) | |
| Prosthetics | Limb reduction defects | Fit early to encourage use |
7. KEY CLASS DISCUSSION POINTS
-
Why does timing matter? — The growing skeleton has remodeling potential unavailable after skeletal maturity. DDH treated at birth with harness avoids surgery; treated at 5 years requires complex osteotomy.
-
Flexible vs. rigid deformity — Always assess passively. Flexible = deformation (positional); corrects with stretching. Rigid = true malformation; requires casting/surgery.
-
Don't miss associations — Congenital scoliosis → VACTERL screen + MRI spine before any surgery. Radial hemimelia → rule out Fanconi anemia (fatal if missed).
-
Neonatal hip examination is a clinical skill — Ortolani and Barlow tests must be performed by every clinician. They become unreliable after 6–8 weeks as soft tissues tighten.
-
Ponseti has changed everything — Before 1990s, extensive posteromedial surgical release was standard for clubfoot, with high rates of stiffness and arthritic feet. Ponseti (non-surgical casting) now achieves > 95% correction with excellent long-term outcomes globally.
-
OI vs. NAI — Every child presenting with multiple unexplained fractures requires multidisciplinary assessment. Blue sclerae, wormian bones, and family history support OI; however, both can coexist.
-
Genetic counseling — Essential for families. Achondroplasia (> 80% de novo), OI (AD with variable expressivity), multifactorial conditions (recurrence risk counseling for DDH, clubfoot).
Sources: Bailey & Love's Short Practice of Surgery, 28th Edition (p. 636, p. 759); Harrison's Principles of Internal Medicine, 21st Edition; AAOS Guidelines — Detection and Nonoperative Management of Pediatric DDH in Infants up to Six Months of Age; ROCO Radiology Dataset; PMC Clinical VQA Dataset; Standard pediatric orthopaedic knowledge base.