porphyria
Reading File
Finding Sources
Searching PubMed
porphyria
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading File
I have comprehensive information. Let me now compile the full answer.
Porphyria
The porphyrias are a group of inherited (occasionally acquired) metabolic disorders caused by defects at specific steps of the heme biosynthetic pathway. Each type is defined by the enzyme deficient, the pattern of intermediate accumulation, and the resulting clinical syndrome. The name derives from the Greek for "purple," reflecting the red-blue discoloration of urine seen in several forms.
The Heme Biosynthetic Pathway
The pathway runs from Glycine + Succinyl-CoA through 8 enzymatic steps to produce heme. It begins and ends in the mitochondrion, with intermediate steps in the cytoplasm. Deficiency of each enzyme produces a distinct disease:
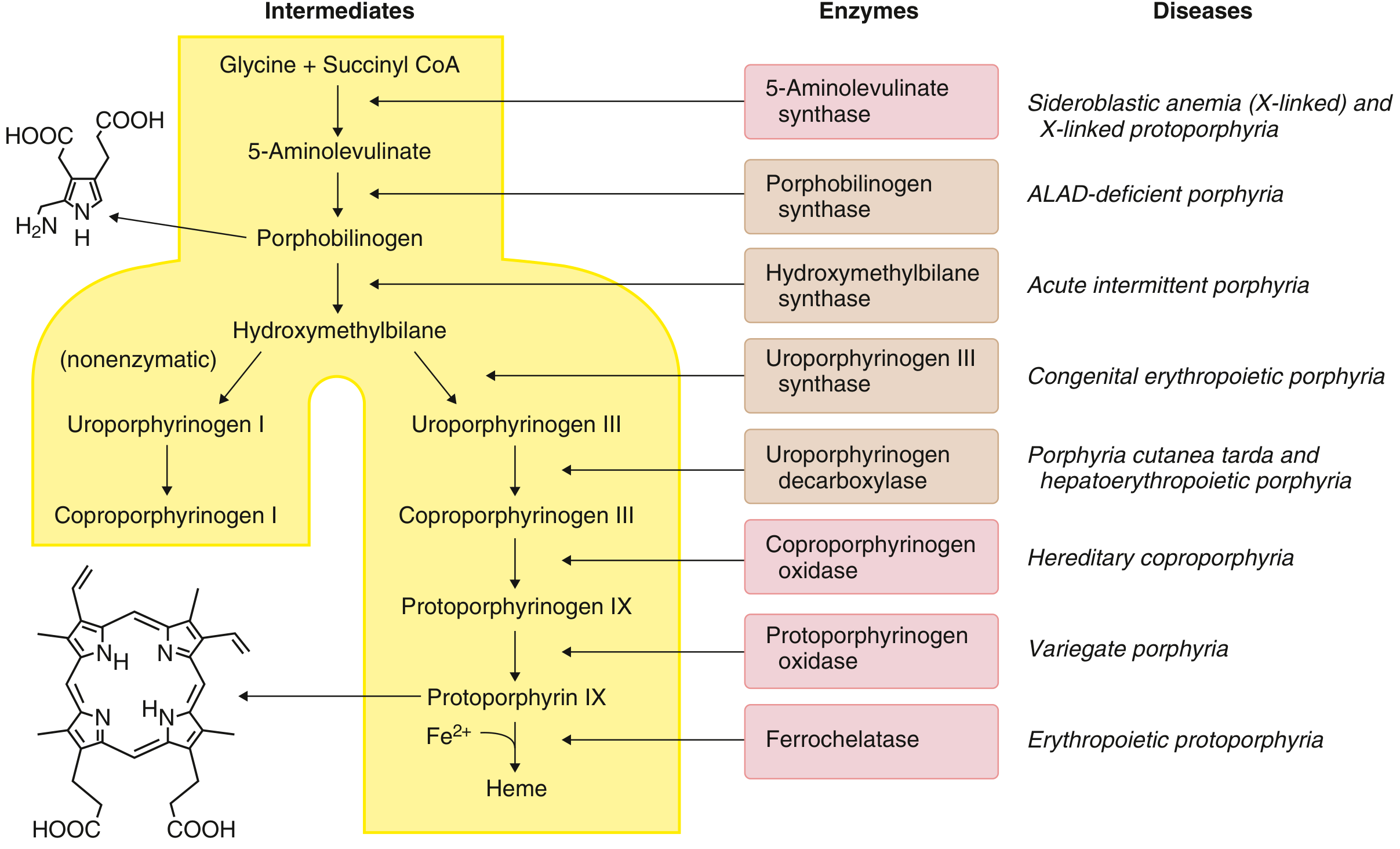
The heme biosynthetic pathway - Goldman-Cecil Medicine
| Step | Enzyme | Disease |
|---|---|---|
| 1 | 5-Aminolevulinate synthase (ALAS) | X-linked sideroblastic anemia / X-linked protoporphyria |
| 2 | Porphobilinogen synthase (ALA dehydratase) | ALAD-deficiency porphyria |
| 3 | Hydroxymethylbilane synthase (PBG deaminase) | Acute intermittent porphyria (AIP) |
| 4 | Uroporphyrinogen III synthase (UROS) | Congenital erythropoietic porphyria (CEP) |
| 5 | Uroporphyrinogen decarboxylase (UROD) | Porphyria cutanea tarda (PCT) / Hepatoerythropoietic porphyria |
| 6 | Coproporphyrinogen oxidase (CPOX) | Hereditary coproporphyria (HCP) |
| 7 | Protoporphyrinogen oxidase (PPOX) | Variegate porphyria (VP) |
| 8 | Ferrochelatase (FECH) | Erythropoietic protoporphyria (EPP) |
Classification
Porphyrias divide into two major groups based on where the enzyme defect occurs:
Hepatic porphyrias - enzyme defect is in the liver
- Acute: AIP, HCP, VP, ALAD-deficiency porphyria
- Chronic: PCT, hepatoerythropoietic porphyria
Erythropoietic porphyrias - enzyme defect is in erythroid precursors of the bone marrow
- Congenital erythropoietic porphyria (CEP)
- Erythropoietic protoporphyria (EPP)
A key clinical principle: enzyme defects before tetrapyrrole formation (i.e., affecting ALA and PBG) cause neurovisceral/acute attacks. Enzyme defects causing accumulation of tetrapyrrolic intermediates (porphyrins) cause photosensitivity, because porphyrins absorb light and generate superoxide radicals that damage membranes. - Lippincott Biochemistry, 8th ed.
Acute Hepatic Porphyrias
Three autosomal dominant forms are clinically important - AIP, HCP, and VP. All share the same acute neurovisceral attack, with some distinctions.
Acute Intermittent Porphyria (AIP)
Enzyme defect: Porphobilinogen deaminase (hydroxymethylbilane synthase)
Genetics: Autosomal dominant
Clinical features of an acute attack:
- Prodrome: Anxiety, restlessness, mild behavioral disturbance
- Established attack: Severe generalized abdominal pain (requires opioids), autonomic overactivity - hypertension, tachycardia, vomiting, constipation, urinary retention
- Warning signs: Weakness of dorsiflexion at wrists and ankles, pain spreading to limbs, worsening hyponatremia
- Critical attack: Rapid-onset motor neuropathy that can progress to quadriplegia and respiratory paralysis; encephalopathy with seizures, confusion, and posterior reversible encephalopathy syndrome (PRES)
No skin disease in AIP (unlike HCP and VP, which can have photosensitivity).
Precipitants: Drugs metabolized by CYP450 (barbiturates, sulfonamides, rifampicin, many anticonvulsants), hormonal changes (pregnancy, menstrual cycle), caloric restriction, alcohol, infection.
Investigations:
- Urine: elevated ALA, PBG, uroporphyrinogen - urine may appear dark/brownish
- CSF protein: normal or mildly elevated
- Hyponatremia (SIADH)
- EDx: markedly reduced CMAP amplitudes, active axonal degeneration on EMG (mimics GBS)
Hereditary Coproporphyria (HCP)
Enzyme defect: Coproporphyrinogen oxidase
Genetics: Autosomal dominant
Causes acute attacks similar to AIP. About one-third of patients also have photosensitive blistering. Fecal coproporphyrin III is always elevated; urinary ALA and PBG rise only during attacks. Plasma fluorescence at 619 nm.
Genetics: Autosomal dominant
Causes acute attacks similar to AIP. About one-third of patients also have photosensitive blistering. Fecal coproporphyrin III is always elevated; urinary ALA and PBG rise only during attacks. Plasma fluorescence at 619 nm.
Variegate Porphyria (VP)
Enzyme defect: Protoporphyrinogen oxidase (PPOX)
Genetics: Autosomal dominant; high penetrance; particularly common in South Africans (founder effect)
Genetics: Autosomal dominant; high penetrance; particularly common in South Africans (founder effect)
VP is the "dual" porphyria - it combines skin lesions of PCT with the acute neurovisceral attacks of AIP. 40-70% have skin symptoms; 27% have acute attacks. Clinical features include blistering and erosions on sun-exposed areas, hypertrichosis, hyperpigmentation, and facial scarring.
Key diagnostic finding: Plasma fluorescence peak at 626 nm - characteristic of VP and distinguishes it from all other porphyrias.
Fecal protoporphyrins and coproporphyrins are always elevated. Urinary coproporphyrins exceed uroporphyrins (distinguishing VP from PCT). - Andrews' Diseases of the Skin
Porphyria Cutanea Tarda (PCT)
Most common porphyria. Caused by deficiency of uroporphyrinogen decarboxylase (UROD).
In 75% of cases, the UROD gene is normal - symptoms result from inhibition of UROD by uroporphomethene (an oxidative product) in the setting of precipitating factors. Only 25% have an inherited (familial) UROD mutation.
Precipitants/associations:
- Hepatic iron overload (HFE gene C282Y mutation is overrepresented)
- Alcohol
- Hepatitis C virus (most common viral association)
- HIV
- Estrogen therapy
- Renal failure, lymphoma, SLE
- Toxins (hexachlorobenzene)
Clinical features: Skin fragility and blistering on sun-exposed areas (especially dorsum of hands), milia, hypertrichosis, pigmentation, sclerodermoid changes. Urine is red-brown in natural light, pink-red in fluorescent light.
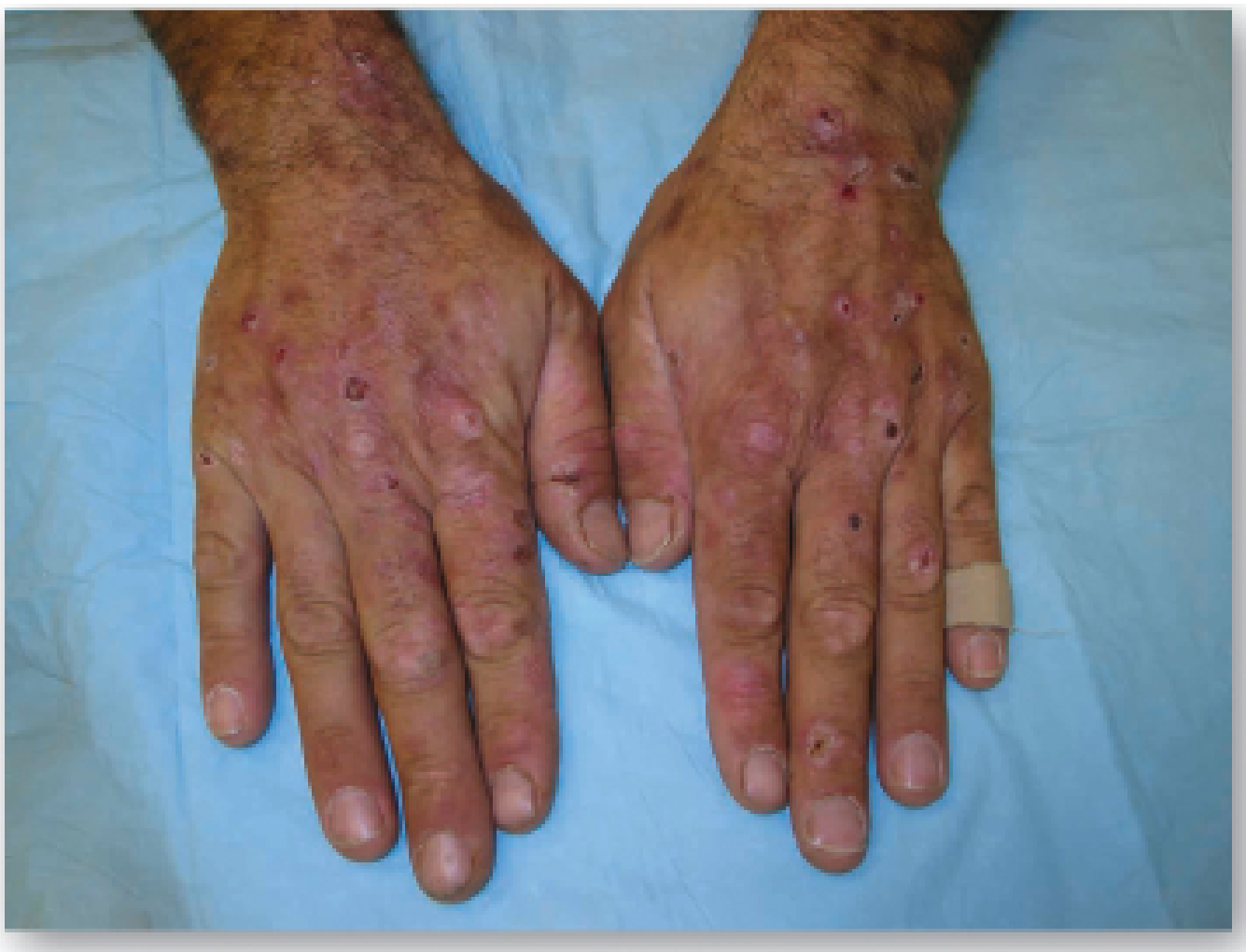
Skin eruptions in PCT - Lippincott Biochemistry
Treatment: Phlebotomy (to reduce hepatic iron) and low-dose hydroxychloroquine/chloroquine (mobilizes porphyrins from liver). Trigger avoidance (alcohol, estrogens, iron supplements). - Goldman-Cecil Medicine
Erythropoietic Protoporphyria (EPP)
Enzyme defect: Ferrochelatase (FECH)
Genetics: Pseudodominant - usually requires one FECH mutation plus a hypomorphic low-expression allele on the other chromosome (found in up to 10-15% of Europeans). Truly autosomal recessive EPP (two loss-of-function mutations) is more severe.
Genetics: Pseudodominant - usually requires one FECH mutation plus a hypomorphic low-expression allele on the other chromosome (found in up to 10-15% of Europeans). Truly autosomal recessive EPP (two loss-of-function mutations) is more severe.
Clinical: Presents in early childhood with painful photosensitivity - burning, erythema, and edema within minutes of sunlight exposure. No blistering (unlike PCT). Over time, waxy scarring and thickening of skin over nose, knuckles. Elevated protoporphyrins in RBCs, plasma, and stool. A minority develop liver disease from protoporphyrin accumulation.
Hepatoerythropoietic Porphyria (HEP)
The homozygous form of PCT. Homozygous/compound heterozygous UROD deficiency (~10% of normal activity). Clinically resembles congenital erythropoietic porphyria (CEP) - dark urine from birth, severe blistering, sclerodermoid scarring, hypertrichosis, red fluorescence of teeth under Wood's light. May require bone marrow transplantation. - Andrews' Diseases of the Skin
Congenital Erythropoietic Porphyria (CEP)
Enzyme defect: Uroporphyrinogen III synthase (UROS)
Genetics: Autosomal recessive (the rarest and most severe erythropoietic porphyria)
Accumulation of uroporphyrinogen I (non-physiological isomer). Severe photomutilation, hemolytic anemia, splenomegaly, hypertrichosis. Severe cases require BMT.
Genetics: Autosomal recessive (the rarest and most severe erythropoietic porphyria)
Accumulation of uroporphyrinogen I (non-physiological isomer). Severe photomutilation, hemolytic anemia, splenomegaly, hypertrichosis. Severe cases require BMT.
Treatment of Acute Attacks
| Treatment | Details |
|---|---|
| IV glucose | 10-20 g/hour - suppresses ALAS1, reducing heme precursor synthesis |
| IV hemin (hematin) | 2-5 mg/kg/day x 3-14 days if no improvement in 24h; directly replenishes heme, down-regulates ALAS1 |
| Givosiran | siRNA (2.5 mg/kg SC monthly) that silences hepatic ALAS1 mRNA - for recurrent AIP. Reduces attack frequency, urinary ALA/PBG, and hemin use - Harrison's 22E |
| GnRH analogs | For premenstrual attacks in VP/AIP |
| Opioid analgesia | For pain management during attacks |
| Avoid triggers | Withdraw causative drugs, maintain caloric intake |
Liver transplantation is curative for recurrent acute hepatic porphyria by removing the source of abnormal ALAS1 expression.
Monitoring and Long-term Risks
- All acute porphyrias (AIP, VP, HCP) confer increased risk of hepatocellular carcinoma (HCC) - a 2024 meta-analysis (PMID 39438413) confirmed this association in acute hepatic porphyria. Regular liver imaging is recommended after age 50.
- Hypertension and chronic kidney disease are common long-term complications of AIP.
- Patients and unaffected gene-carrier relatives must be educated about porphyrogenic drugs and lifestyle triggers.
Quick Reference - Key Distinguishing Features
| Feature | AIP | VP | HCP | PCT | EPP |
|---|---|---|---|---|---|
| Acute attacks | + | + (27%) | + (35%) | - | - |
| Photosensitivity | - | + (40-70%) | + (33%) | + | + |
| Enzyme | HMBS | PPOX | CPOX | UROD | FECH |
| Inheritance | AD | AD | AD | Mostly sporadic | Pseudodominant |
| Diagnostic marker | Urine PBG↑↑ | Plasma 626 nm | Fecal copro III↑ | Urine uroporphyrin↑ | RBC protoporphyrin↑ |
| Treatment | Hemin/glucose/givosiran | Hemin/glucose | Hemin/glucose | Phlebotomy/chloroquine | Sun protection/afamelanotide |
Sources: Goldman-Cecil Medicine 2-Vol Set; Harrison's Principles of Internal Medicine 22E; Lippincott Illustrated Reviews Biochemistry 8th ed; Andrews' Diseases of the Skin; Bradley and Daroff's Neurology in Clinical Practice.
This is a shared conversation. Sign in to Orris to start your own chat.