Topic 2) basic adrenal anatomy,histology and overview Topic 3) adrenal cortex:Cushing syndrome Topic 4) adrenal cortical insufficiency Topic 5) neoplasm of adrenal cortex Topic 6)adrenal medullary neoplasm
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive, structured study guide covering all five topics drawn from Schwartz's Principles of Surgery, Campbell-Walsh Urology, and Brenner & Rector's The Kidney.
ADRENAL GLAND - Comprehensive Study Notes (Topics 2-6)
TOPIC 2: Basic Adrenal Anatomy, Histology & Overview
Historical Background
- Eustachius gave the first anatomic description of the adrenals in 1563
- Cuvier described the cortex-medulla division in 1805
- Thomas Addison described adrenal insufficiency in 1855
- Harvey Cushing described his syndrome in 1932 (11 patients with moon facies, truncal obesity, hypertension)
- Aldosterone was identified in 1952; Conn's syndrome described in 1955
Embryology
The adrenal gland is essentially two organs in one, with entirely distinct origins:
| Component | Embryologic Origin | Timing |
|---|---|---|
| Cortex | Mesoderm (adrenogenital ridge) | ~5th week of gestation |
| Medulla | Neuroectoderm (neural crest cells) | ~5th week; migrates to cortex |
Key points:
- The fetal cortex is functional by 8 weeks and produces fetal steroids, but involutes after birth - adrenal weight decreases in first 3 postpartum months
- The definitive cortex forms the adult cortex over the first 3 years of life
- Because the cortex originates near the gonads, ectopic adrenocortical tissue can be found in the ovaries, spermatic cord, and testes
- Neural crest cells migrate to form the medulla; most extra-adrenal neural tissue regresses, but the organ of Zuckerkandl (near the aortic bifurcation/inferior mesenteric artery origin) is the largest persistent remnant
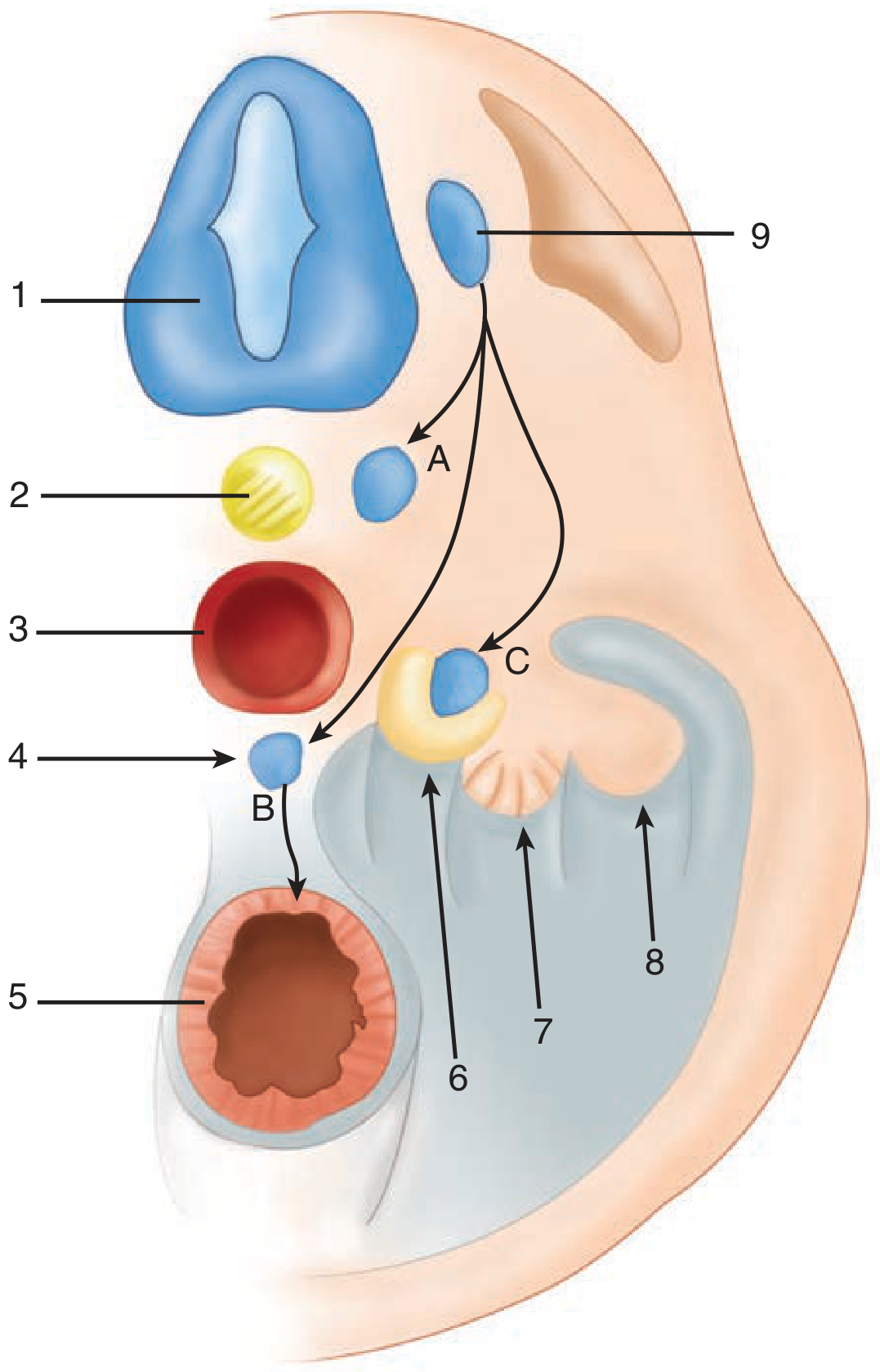
Figure: Cross-section of the embryo depicting adrenal development - Schwartz's Principles of Surgery, 11e
Adult Anatomy
- Right adrenal: pyramidal shape, sits above the right kidney; right adrenal vein drains directly into the inferior vena cava (short and wide - high risk of hemorrhage during surgery)
- Left adrenal: semilunar/crescentic shape; left adrenal vein drains into the left renal vein
- Arterial supply: three sources - superior suprarenal artery (from inferior phrenic), middle suprarenal (directly from aorta), inferior suprarenal (from renal artery)
- Weight: each gland ~4-5 g in adults
Histology - The Three Zones of the Cortex
The cortex is divided into three concentric zones ("GFR" = from outer to inner):
| Zone | Mnemonic | Product | Regulation |
|---|---|---|---|
| Zona Glomerulosa (outermost) | "Salt" | Mineralocorticoids (aldosterone) | Renin-angiotensin system, K+ |
| Zona Fasciculata (middle, largest) | "Sugar" | Glucocorticoids (cortisol) | ACTH |
| Zona Reticularis (innermost) | "Sex" | Androgens (DHEA, androstenedione) | ACTH |
Memory aid: "GFR" - Glomerulosa, Fasciculata, Reticularis (outer to inner); products: "Salt, Sugar, Sex"
- The medulla contains chromaffin cells (modified postganglionic sympathetic neurons) that produce catecholamines: epinephrine (~80%) and norepinephrine (~20%)
- Medullary cells stain brown with chromate salts - the "chromaffin reaction"
Adrenal Physiology Overview
- Cortisol: zona fasciculata secretes up to 20 mg/day; follows a circadian rhythm - peak in morning, nadir at ~11 PM
- HPA Axis: Hypothalamus → CRH → Anterior pituitary → ACTH → Adrenal cortex → Cortisol → negative feedback
- ACTH is cleaved from the precursor molecule pro-opiomelanocortin (POMC)
- Without ACTH, all cortical cells except mineralocorticoid-producing zona glomerulosa undergo apoptosis
TOPIC 3: Adrenal Cortex - Cushing Syndrome
Definition & Epidemiology
- Cushing syndrome: hypercortisolism from excessive glucocorticoid production
- Incidence: 2-5 per million per year (rare)
- Most common cause today: exogenous/iatrogenic (prescribed corticosteroids)
Causes - Classification
| Category | Cause | % of Endogenous Cases |
|---|---|---|
| ACTH-dependent | Cushing's disease (pituitary adenoma) | ~60-70% |
| ACTH-dependent | Ectopic ACTH (small cell lung ca, bronchial carcinoid) | ~10-12% |
| ACTH-independent | Adrenal adenoma | ~10-15% |
| ACTH-independent | Adrenal carcinoma | ~5-10% |
| ACTH-independent | AIMAH (bilateral macronodular hyperplasia) | rare |
Key: Adrenal tumors secreting cortisol are ACTH-independent and account for ~10% of non-iatrogenic cases.
Clinical Features
| System | Manifestation |
|---|---|
| General | Central obesity, buffalo hump, moon facies, supraclavicular fat pads |
| Skin | Hirsutism, plethora, purple striae, acne, easy bruising, skin atrophy |
| Cardiovascular | Hypertension |
| Musculoskeletal | Proximal muscle weakness, osteopenia/osteoporosis |
| Metabolic | Diabetes/glucose intolerance, hyperlipidemia |
| Neuropsychiatric | Depression, emotional lability, psychosis |
| Renal | Polyuria, renal stones |
| Gonadal | Impotence, decreased libido, menstrual irregularities |
- Hyperpigmentation suggests ectopic ACTH (very high circulating ACTH levels stimulate melanocortin receptors)
- Children: obesity + stunted growth (growth suppression distinguishes it from simple obesity)
Diagnostic Workup
Step 1 - Confirm hypercortisolism (screening):
- Overnight low-dose DST: 1 mg dexamethasone at 11 PM → cortisol at 8 AM. Normal suppresses to <3 μg/dL (or <1.8 μg/dL by strict criteria)
- 24-hour urinary free cortisol: sensitivity 95-100%, specificity 98%; <100 μg/day rules out hypercortisolism
- Late-night salivary cortisol: reflects loss of diurnal rhythm; increasingly used
Step 2 - Determine ACTH dependence:
- Plasma ACTH levels (normal 10-100 pg/mL):
- Cushing's disease: 15-500 pg/mL (elevated)
- Ectopic ACTH: >1000 pg/mL (markedly elevated)
- Adrenal tumor: <5 pg/mL (suppressed)
Step 3 - Localize:
- High-dose DST (2 mg q6h x 8 doses, or 8 mg overnight): pituitary adenoma usually suppresses; ectopic ACTH does not
- CRH stimulation test: pituitary adenoma responds (ACTH rises); ectopic ACTH does not
- MRI pituitary for Cushing's disease
- CT adrenal for adrenal cause
- Petrosal sinus sampling: gold standard to distinguish pituitary vs. ectopic when imaging inconclusive
Treatment
- Cushing's disease: trans-sphenoidal pituitary surgery (first line)
- Adrenal adenoma: laparoscopic adrenalectomy (adrenal-dependent disease)
- Bilateral adrenal disease / ectopic ACTH not resectable: bilateral adrenalectomy
- Medical bridging: metyrapone, ketoconazole, aminoglutethimide, etomidate (block steroidogenesis); mitotane
- Prognosis: life expectancy reduced; cardiovascular risk persists even after normalization of cortisol
Subclinical Cushing Syndrome
- Occurs in ~8% of adrenal incidentalomas
- No overt clinical features, but subtle cortisol excess (loss of diurnal variation, resistance to dexamethasone suppression)
- Risk: postoperative adrenal crisis due to unrecognized contralateral adrenal suppression
TOPIC 4: Adrenal Cortical Insufficiency (Addison's Disease)
Classification
| Type | Mechanism | Examples |
|---|---|---|
| Primary (Addison's disease) | Direct adrenal destruction | Autoimmune (most common in developed world), TB (most common worldwide historically), fungal infection, metastases, hemorrhage, sarcoidosis |
| Secondary | Insufficient ACTH | Pituitary disease, exogenous steroid withdrawal |
| Tertiary | Insufficient CRH | Hypothalamic disease |
Primary vs. Secondary: Key Differences
| Feature | Primary (Addison's) | Secondary |
|---|---|---|
| Aldosterone | Deficient (mineralocorticoid deficit) | Normal (RAAS intact) |
| ACTH | Very HIGH (loss of feedback) | Low |
| Skin pigmentation | YES - hyperpigmentation (high ACTH/MSH) | No |
| Hyponatremia | Hyponatremia + hyperkalemia | Hyponatremia only (no hyperkalemia) |
Clinical Features of Primary Adrenal Insufficiency
- Chronic: fatigue, weakness, anorexia, weight loss, nausea, hyperpigmentation (buccal mucosa, scars, creases), salt craving, postural hypotension
- Electrolytes: hyponatremia, hyperkalemia, mild acidosis (from aldosterone deficiency)
- Adrenal crisis (acute): severe hypotension/shock, vomiting, abdominal pain, fever, hypoglycemia - precipitated by stress (infection, trauma, surgery)
Diagnosis
- 8 AM cortisol: <3 μg/dL is diagnostic; >18 μg/dL excludes
- ACTH stimulation test (cosyntropin test): give 250 μg synthetic ACTH IV; cortisol should rise to >18 μg/dL. Failure confirms adrenal insufficiency
- Plasma ACTH: elevated in primary, low in secondary
- Anti-21-hydroxylase antibodies: positive in ~90% of autoimmune Addison's
Treatment
- Chronic replacement:
- Hydrocortisone 15-25 mg/day (split doses; higher in morning to mimic diurnal rhythm)
- Fludrocortisone 0.05-0.2 mg/day (mineralocorticoid replacement - only needed in primary)
- DHEA replacement in some patients (especially women, for well-being/libido)
- Sick day rules: double/triple hydrocortisone dose during illness
- Adrenal crisis: IV hydrocortisone 100 mg bolus → 50-100 mg q6-8h + aggressive IV fluids (normal saline + dextrose)
Congenital Adrenal Hyperplasia (CAH)
- Autosomal recessive deficiency of steroidogenic enzymes
- Most common: 21-hydroxylase deficiency (~90%) → cannot make cortisol → excess ACTH → adrenal hyperplasia + excess androgens
- Classic forms: salt-wasting (severe, neonatal crisis) and simple virilizing
- Results in female pseudohermaphroditism (virilization of 46,XX females)
TOPIC 5: Neoplasms of the Adrenal Cortex
Adrenal Adenoma
- Benign, usually functioning (Conn's or Cushing's) or non-functioning
- Imaging (CT): homogeneous, well-encapsulated, smooth margins, <10 Hounsfield units (lipid-rich), low attenuation
- MRI: low signal intensity T2-weighted relative to liver (mass-to-liver ratio <1.4)
- Most are <4 cm; carcinoma accounts for only 2% of lesions <4 cm
Adrenocortical Carcinoma (ACC)
- Rare, highly aggressive malignancy
- Incidence: ~1-2 per million per year
- Most present with large tumors (median 10-12 cm at diagnosis)
- ~60% are functioning - most commonly excess cortisol ± androgens (Cushing + virilization is a suspicious combination)
Imaging features suggesting malignancy:
- Size >6 cm (~25% malignancy risk; but carcinomas occur in smaller lesions too)
- CT attenuation >18 Hounsfield units (lipid-poor)
- Inhomogeneous, irregular borders, local invasion, lymphadenopathy
- MRI T2: moderate-high signal intensity (mass-to-liver ratio 1.2-2.8)
- Slow washout of contrast on delayed CT imaging
Staging (modified Sullivan/Macfarlane):
- Stage I: <5 cm, no invasion
- Stage II: >5 cm, no invasion
- Stage III: local invasion or nodal disease
- Stage IV: distant metastases
Treatment:
- Complete surgical resection (open adrenalectomy preferred for ACC - wide margins needed)
- Mitotane (adrenolytic agent): adjuvant treatment; also used for unresectable/metastatic disease
- Additional chemotherapy: EDP-M regimen (etoposide, doxorubicin, cisplatin + mitotane)
- Prognosis is poor: 5-year survival ~15-35% overall
Adrenal Incidentaloma
- Adrenal mass found incidentally on imaging performed for another reason
- Prevalence increases with age (~5% of abdominal CT scans)
- Mandatory workup for ALL incidentalomas (except obvious cysts, hemorrhage, myelolipoma, diffuse mets):
- Overnight 1 mg DST (rule out subclinical Cushing)
- 24-hour urine catecholamines/metanephrines OR plasma metanephrines (rule out pheochromocytoma - must exclude before any procedure)
- Aldosterone/renin ratio if hypertensive (rule out Conn's)
- 17-ketosteroids if sex steroid excess suspected
Indications for adrenalectomy:
- Functioning tumor (any hormone excess)
- Size >4-6 cm (increased malignancy risk)
- Growth >1 cm on follow-up imaging
Primary Aldosteronism (Conn's Syndrome)
- Most common cause of secondary hypertension (3-13% of hypertensive patients in primary care; up to 30% at referral centers)
- Characterized by autonomous aldosterone production → suppressed renin → hypertension + hypokalemia (though hypokalemia absent in ~50%)
- Subtypes: unilateral adrenal adenoma (~35-40%) vs. bilateral idiopathic hyperplasia (~60%)
- Diagnosis: aldosterone-to-renin ratio (ARR) as screening test
- Treatment: adenoma → laparoscopic adrenalectomy; bilateral hyperplasia → mineralocorticoid antagonist (spironolactone/eplerenone)
Adrenal Myelolipoma
- Benign, non-functioning lesion
- Composed of hematopoietic tissue + mature adipose tissue
- Characteristic CT appearance: fat attenuation (negative Hounsfield units)
- Usually requires no treatment; surgery if symptomatic or large
TOPIC 6: Adrenal Medullary Neoplasms
Pheochromocytoma
Definition & Epidemiology
- Catecholamine-secreting tumor arising from chromaffin cells of the adrenal medulla
- Estimated incidence: 2-8 cases per million per year
- Found in 0.2-0.6% of hypertensive patients (1.7% of hypertensive children)
- Paraganglioma: same tumor type arising from extra-adrenal chromaffin tissue (sympathetic paraganglia)
The "10% Rule" (classic, now updated)
| Feature | Classic "10%" | Modern reality |
|---|---|---|
| Extra-adrenal | ~10% | ~10-15% |
| Bilateral | ~10% | Higher in familial cases |
| Malignant | ~10% | >10% metastatic at diagnosis |
| Familial | ~10% | Now >30% have germline mutations |
| In children | ~10% | More common than thought |
Familial Syndromes Involving Pheochromocytoma
| Syndrome | Gene | Associated Features |
|---|---|---|
| MEN 2A | RET proto-oncogene | Pheo (~50%, often bilateral), medullary thyroid carcinoma, parathyroid adenoma, cutaneous lichen amyloidosis |
| MEN 2B | RET | Pheo (usually bilateral), medullary thyroid ca, mucosal neuromas, marfanoid habitus, Hirschsprung disease |
| Von Hippel-Lindau type 2 | VHL (3p25-26) | ~20% get pheo/paraganglioma; renal cell carcinoma, cerebellar hemangioblastoma, retinal angiomas |
| Neurofibromatosis type 1 | NF1 (17q11.2) | ~2% get catecholamine-secreting tumor; café-au-lait spots, neurofibromas, Lisch nodules |
| SDH mutations | SDHB, SDHC, SDHD | Paraganglioma syndromes; SDHB strongly associated with malignancy |
Clinical Features
- Classic triad: episodic headache, diaphoresis (sweating), and hypertension (sustained or paroxysmal)
- Present in 95% of cases in some large series
- Other: palpitations, pallor, anxiety, weight loss, hyperglycemia
- Hypertensive crisis can be fatal and may be precipitated by surgery, anesthesia, contrast dye, certain medications (beta-blockers, TCA, metoclopramide)
- May present as: heart failure, cardiomyopathy, stroke, or incidentaloma
Diagnosis
Biochemical (first step):
- Plasma free metanephrines (normetanephrine + metanephrine): highest sensitivity (~99%); test of choice for screening
- 24-hour urine catecholamines + metanephrines + VMA: good sensitivity, widely available
- Combination of plasma + urine tests maximizes accuracy
Localization (after biochemical confirmation):
- CT abdomen/pelvis: first-line imaging; 90-95% sensitivity for adrenal pheo
- MRI: preferred in children, pregnancy, known metastases; pheochromocytoma is characteristically very bright on T2 (mass-to-liver ratio >3)
- MIBG scan (metaiodobenzylguanidine): functional imaging; useful for extra-adrenal, metastatic, and recurrent disease
- PET scan (FDOPA or FDG-PET): increasingly used for metastatic/familial disease
Preoperative Preparation (CRITICAL)
- Alpha-blockade FIRST (phenoxybenzamine 10-40 mg/day, titrated over 7-14 days; or prazosin/doxazosin)
- Beta-blockade only AFTER adequate alpha-blockade (to control tachycardia) - NEVER give beta-blocker first (risks unopposed alpha causing hypertensive crisis)
- Liberal salt and fluid intake to expand contracted intravascular volume
- Delay surgery 7-14 days after localization to normalize BP, heart rate, and volume
- Metyrosine (tyrosine hydroxylase inhibitor) for very large tumors or non-surgical candidates
Surgery
- Laparoscopic adrenalectomy: preferred for small, benign-appearing adrenal pheochromocytomas
- Open approach: for large tumors, suspected malignancy, or paragangliomas in difficult locations
- Intraoperative risks: severe hypertension during tumor manipulation, severe hypotension after ligation of adrenal vein
- Postoperative: monitor for hypotension, hypoglycemia, adrenal insufficiency
Follow-up
- Repeat biochemical testing 4-6 weeks postop (to confirm complete resection)
- Long-term annual follow-up (recurrence, metachronous tumors, especially in familial cases)
- Genetic testing recommended for all patients - especially for SDHB (high malignancy risk), VHL, RET, NF1
Malignant Pheochromocytoma
- Defined by presence of metastases (not histologic features alone) in sites where chromaffin tissue is normally absent (bone, liver, lung, lymph nodes)
-
10% are malignant at diagnosis; SDHB mutation is the strongest predictor
- Treatment: surgery + MIBG therapy, sunitinib, CVD chemotherapy (cyclophosphamide-vincristine-dacarbazine)
- Prognosis: highly variable; 5-year survival ~50% for malignant disease
Neuroblastoma
- Most common extracranial solid tumor of childhood
- Arises from neural crest cells (same as pheochromocytoma) - can arise in the adrenal medulla or anywhere along the sympathetic chain
- Secretes catecholamines (mostly norepinephrine + dopamine) but rarely causes hypertension
- Presents with abdominal mass, opsoclonus-myoclonus, skin nodules ("blueberry muffin"), bone pain, orbital ecchymosis ("raccoon eyes")
- Biochemical marker: elevated urine VMA and HVA (vanillylmandelic acid and homovanillic acid)
- Genetics: MYCN amplification (poor prognosis), chromosome 1p deletion
- Treatment: surgery, chemotherapy, radiation, differentiation therapy (retinoic acid), MIBG therapy
Quick Comparison Summary
| Cushing | Conn's | Addison's | ACC | Pheo | |
|---|---|---|---|---|---|
| Hormone excess | Cortisol | Aldosterone | Deficiency (cortisol + aldosterone) | Cortisol ± androgens | Catecholamines |
| BP | High | High | Low (crisis) | High (if functional) | Paroxysmal/sustained high |
| K+ | Normal/low | Low | High | Low | Normal |
| Na+ | Normal/high | High/normal | Low | - | Normal |
| ACTH | Variable | Normal | Very HIGH (primary) | Suppressed | Normal |
| Key test | DST/24-hr UFC | ARR | Cosyntropin stim | CT/imaging | Plasma metanephrines |
| Treatment | Surgery/medical | Adrenalectomy/spironolactone | Hydrocortisone + fludrocortisone | Open resection + mitotane | Alpha-block THEN surgery |
Sources: Schwartz's Principles of Surgery 11e, Campbell-Walsh Urology (9780323546423), Brenner & Rector's The Kidney
Acth and cry full form
ACTH = Adrenocorticotropic Hormone (also called corticotropin)
CRH = Corticotropin-Releasing Hormone (produced by the hypothalamus)
These are part of the HPA axis:
Hypothalamus → releases CRH → Anterior Pituitary → releases ACTH → Adrenal Cortex → releases Cortisol
This is a shared conversation. Sign in to Orris to start your own chat.