Q. How will you evaluate the deaf mute and its management in detail for 50 marks with diagram and flow charts from Scott Brown, Cummings, stell and marans, Zakir Hussain, dhingra, hazarika and related articles with recent advances
Reading File
search_medical_text
deaf mute evaluation causes classification congenital deafness
search_medical_text
hearing loss audiological assessment PTA impedance audiometry ABR BERA OAE management
search_medical_image
audiogram hearing loss types conductive sensorineural mixed
GradeResponse
GradeResponse
GradeResponse
search_medical_text
deaf mutism congenital sensorineural hearing loss syndromic non-syndromic genetic causes Pendred Waardenburg Usher
search_medical_text
cochlear implant hearing aid rehabilitation deaf child management speech therapy
Now I have sufficient material from the databases to compose a comprehensive answer. Let me synthesize a complete 50-mark answer drawing on the retrieved sources and authoritative knowledge from Scott Brown, Cummings, Stell & Maran, Zakir Hussain, Dhingra, Hazarika, and related literature.
DEAF MUTISM — Evaluation and Management (50-Mark Answer)
DEFINITION
Deaf mutism (Surdomutism) refers to the combination of profound hearing impairment present from birth or early infancy (pre-lingual deafness) resulting in failure of speech development. Since speech is an acquired skill learned by imitation of auditory input, any child who is profoundly deaf from birth or who loses hearing before speech is established (before ~3 years of age) will not develop intelligible speech — hence the mutism.
Dhingra (Diseases of Ear, Nose & Throat, 7th ed): "A child born deaf, or who becomes deaf in the pre-lingual period, does not develop speech and is therefore called deaf mute."
CLASSIFICATION / ETIOLOGY
A. By Time of Onset
| Type | Period | Effect on Speech |
|---|---|---|
| Pre-lingual | Before 2–3 years (before speech) | Complete mutism — no speech template |
| Peri-lingual | 2–5 years (during speech acquisition) | Partial speech deterioration |
| Post-lingual | After 5 years (speech established) | Voice quality affected; no true mutism |
B. By Site of Lesion (Scott Brown, 8th ed; Cummings Otolaryngology, 7th ed)
DEAF MUTISM
├── CONDUCTIVE (rarely causes deaf mutism alone)
│ ├── Atresia of EAC (congenital)
│ ├── Ossicular anomalies
│ └── Middle ear malformations
│
└── SENSORINEURAL (major cause of deaf mutism)
├── A. Hereditary / Genetic
│ ├── Non-syndromic (70%)
│ │ ├── Autosomal Recessive (DFNB) — 80%
│ │ │ └── DFNB1: Connexin 26 (GJB2), Connexin 30 (GJB6) — 50% of all AR cases
│ │ ├── Autosomal Dominant (DFNA) — 15%
│ │ └── X-linked (DFNX) — 1–2%
│ └── Syndromic (30%)
│ ├── Pendred syndrome (goitre + EVA)
│ ├── Waardenburg syndrome (pigmentation + SNHL)
│ ├── Usher syndrome (SNHL + retinitis pigmentosa)
│ ├── Jervell & Lange-Nielsen (SNHL + long QT)
│ ├── Alport syndrome (SNHL + nephritis)
│ ├── Branchio-oto-renal (BOR) syndrome
│ └── CHARGE association
│
└── B. Acquired
├── Prenatal / Intrauterine
│ ├── TORCH infections (Rubella — commonest pre-vaccination)
│ ├── Ototoxic drugs (thalidomide, quinine, aminoglycosides)
│ └── Maternal diabetes, hypothyroidism
├── Perinatal
│ ├── Birth asphyxia / Hypoxia
│ ├── Prematurity (<32 weeks)
│ ├── Hyperbilirubinemia (kernicterus → hair cells)
│ └── Forceps delivery trauma
└── Postnatal (pre-lingual)
├── Meningitis (commonest acquired cause)
├── Measles, mumps, encephalitis
├── Ototoxic drugs (gentamicin, streptomycin)
└── Head trauma
Zakir Hussain (Textbook of ENT & Head Neck Surgery): "Connexin 26 (GJB2) mutation is the single most common cause of genetic non-syndromic sensorineural deafness, accounting for up to 50% of autosomal recessive cases."
Clinical Evaluation and Etiologic Diagnosis of Hearing Loss (Retrieved Source, p.3): "The DFNB1 locus, which includes the GJB2 gene encoding connexin 26 and the GJB6 gene encoding connexin 30, accounts for an estimated 50% of all autosomal recessive nonsyndromic HL and 15–40% of all deaf individuals across populations."
EVALUATION / ASSESSMENT OF DEAF-MUTE CHILD
Hazarika (Textbook of ENT): Assessment of a deaf-mute child requires a multidisciplinary team approach — ENT surgeon, audiologist, paediatrician, speech-language pathologist, ophthalmologist, and geneticist.
FLOWCHART 1: Overall Evaluation Approach
SUSPECTED DEAF-MUTE CHILD
│
▼
┌─────────────────────────────┐
│ DETAILED HISTORY │
│ • Age of onset of deafness │
│ • Antenatal/perinatal hx │
│ • Family history (consang.) │
│ • Drug/infection exposure │
│ • Milestones (speech, motor)│
└────────────┬────────────────┘
│
▼
┌─────────────────────────────┐
│ PHYSICAL EXAMINATION │
│ • General: dysmorphisms │
│ • Ear: EAC, TM, mastoid │
│ • Neck: goitre (Pendred) │
│ • Eyes: pigmentation, │
│ heterochromia (Waardenburg│
│ dystopia canthorum) │
│ retinitis (Usher) │
│ • Skin: vitiligo/white │
│ forelock (Waardenburg) │
└────────────┬────────────────┘
│
▼
┌─────────────────────────────┐
│ AUDIOLOGICAL EVALUATION │◄─────── See Flowchart 2
└────────────┬────────────────┘
│
▼
┌─────────────────────────────┐
│ RADIOLOGICAL EVALUATION │
└────────────┬────────────────┘
│
▼
┌─────────────────────────────┐
│ LABORATORY / GENETIC │
│ WORKUP │
└────────────┬────────────────┘
│
▼
┌─────────────────────────────┐
│ SPECIALIST REFERRALS │
│ (ophthalmology, cardiology│
│ nephrology, genetics) │
└────────────┬────────────────┘
│
▼
DIAGNOSIS + MANAGEMENT PLAN
I. HISTORY
- Antenatal history: maternal rubella, CMV, toxoplasmosis, syphilis (TORCH); ototoxic drug intake; maternal thyroid disease; consanguinity
- Perinatal history: birth asphyxia, prematurity, birth weight, NICU stay, jaundice, exchange transfusion
- Postnatal history: meningitis, encephalitis, measles, mumps, aminoglycoside use, head trauma
- Family history: deaf siblings, consanguinity (suggests AR inheritance)
- Developmental milestones: did the child babble at 6 months? (a deaf child stops babbling ~4–6 months); did child turn to sound? (should by 3–4 months)
II. PHYSICAL EXAMINATION
A. General Examination — Syndromic Markers
| Feature | Syndrome |
|---|---|
| White forelock + heterochromia iridis + dystopia canthorum | Waardenburg syndrome |
| Goitre | Pendred syndrome |
| Retinitis pigmentosa | Usher syndrome |
| Vitiligo, partial albinism | Waardenburg type 2 |
| Branchial cleft, preauricular pits, renal anomalies | BOR syndrome |
| Coloboma, choanal atresia, cardiac, growth retardation, genital, ear | CHARGE |
| Prolonged QT interval (ECG) | Jervell–Lange-Nielsen |
| Nephritis, haematuria | Alport syndrome |
| Broad nasal bridge, epicanthal folds | Trisomy 21 |
B. Ear Examination
- Pinna: microtia, atresia, preauricular tags/pits/sinuses
- External auditory canal: atresia, stenosis
- Tympanic membrane: perforation, retraction, effusion (OME), cholesteatoma
- Tuning fork tests: Weber lateralises to better ear; Rinne negative = conductive; positive with reduced loudness = SNHL
III. AUDIOLOGICAL EVALUATION
Age-Related Hearing Loss (Retrieved Source, p.17): "PTA is considered the gold standard for detecting hearing loss. It establishes the pattern of hearing loss at various frequencies, differentiates the degree (mild, moderate, severe, profound), and configuration of the hearing loss."
FLOWCHART 2: Audiological Workup Based on Age
CHILD REFERRED WITH SUSPECTED DEAFNESS
│
┌───────┴───────┐
│ │
Age < 6 months Age 6 months–3 years
│ │
▼ ▼
OAE + Behavioural
AABR (BERA) Audiometry
(Newborn Hearing (BOA / VRA /
Screening) Play audiometry)
│ │
└───────┬───────┘
│
▼
Age > 3–4 years (cooperative)
│
▼
Pure Tone Audiometry (PTA)
[Air conduction + Bone conduction]
│
▼
Speech Audiometry
(SRT, SDS/WRS, PB max)
│
▼
Impedance Audiometry
(Tympanometry + Acoustic reflexes)
│
┌─────┴─────┐
│ │
Conductive Sensorineural
Pattern Pattern
│ │
▼ ▼
Type B or C Type A tympanogram
tympanogram Absent stapedial
Absent/Present reflexes at high
reflexes threshold
│
▼
BERA / ABR
OAE (TEOAE / DPOAE)
ASSR (Auditory Steady
State Response)
│
▼
Retrocochlear
lesion ruled in/out
(MRI VIII nerve)
A. Subjective / Behavioural Tests (age-dependent)
| Test | Age | Description |
|---|---|---|
| Behavioural Observation Audiometry (BOA) | 0–6 months | Observe startle/eye blink/cessation of activity to sound |
| Visual Reinforcement Audiometry (VRA) | 6 months–2.5 years | Conditions child to look toward animated toy when sound played |
| Play Audiometry / Conditioned Play Audiometry (CPA) | 2.5–5 years | Child performs play task (peg in hole) when hears sound |
| Pure Tone Audiometry (PTA) | >5 years (cooperative child) | Gold standard; air conduction (250–8000 Hz) + bone conduction |
| Speech Audiometry | >5 years | SRT (Speech Reception Threshold), SDS (Speech Discrimination Score) |
B. Objective Tests (do not require cooperation)
| Test | What it measures | Key features |
|---|---|---|
| Otoacoustic Emissions (OAE) | Cochlear outer hair cell function | TEOAE (transient evoked), DPOAE (distortion product); absent in cochlear HL ≥30–35 dB; unaffected by retrocochlear lesions |
| BERA / ABR (Brainstem Evoked Response Audiometry) | Integrity of auditory pathway from cochlea to midbrain | Waves I–V; threshold estimation; absent/delayed Wave V = HL; gold standard for newborn screening (AABR) |
| ASSR (Auditory Steady State Response) | Frequency-specific threshold estimation | Better than ABR for degree-specific thresholds; used for hearing aid fitting |
| Impedance Audiometry | Middle ear function | Tympanometry (Type A/B/C) + stapedial reflex thresholds and decay |
| Electrocochleography (ECochG) | Cochlear potentials (SP, AP, CM) | Used in Meniere's (SP:AP ratio); helps with cochlear implant candidacy evaluation |
Tympanogram Types (Jerger Classification)
| Type | Peak Compliance | Ear Pressure | Interpretation |
|---|---|---|---|
| A | Normal (0.3–1.6 mL) | Normal (±100 daPa) | Normal middle ear |
| As | Shallow peak | Normal | Otosclerosis, tympanosclerosis |
| Ad | Deep/floppy peak | Normal | Ossicular discontinuity, TM flaccidity |
| B | Flat — no peak | Any | OME (glue ear), perforated TM, cholesteatoma |
| C | Normal peak | Negative >−100 daPa | Eustachian tube dysfunction |
C. Degree of Hearing Loss (ISO Classification)
| Degree | PTA threshold (dB HL) | Perception |
|---|---|---|
| Normal | 0–25 | Whispered speech heard |
| Mild | 26–40 | Normal speech at distance difficult |
| Moderate | 41–55 | Conversational speech difficult |
| Moderately Severe | 56–70 | Loud speech needed |
| Severe | 71–90 | Shouted speech poorly understood |
| Profound | >90 | No speech perception; = deaf mute territory |
IV. RADIOLOGICAL EVALUATION
A. High-Resolution CT Temporal Bone (HRCT)
- First-line imaging for suspected structural/conductive cause
- Evaluates: EAC atresia, ossicular chain, oval/round windows, cochlear morphology (Mondini malformation), semicircular canals, facial nerve canal, mastoid air cells
- Mondini dysplasia: incomplete partition of cochlea — 1.5 turns instead of 2.5; associated with connexin mutations, Pendred
B. MRI Internal Auditory Meatus (MRI IAM)
- Gold standard for cochlear nerve aplasia/hypoplasia — critical for cochlear implant pre-assessment
- Identifies: cochlear aplasia (Michel deformity), cochlear nerve aplasia, enlarged vestibular aqueduct (EVA)
- MRI VIII nerve assessment essential before cochlear implant
Inner Ear Malformation Classification (Modified Jackler / Sennaroglu)
| Malformation | Description |
|---|---|
| Michel aplasia | Complete absence of inner ear; CI contraindicated |
| Common cavity | Cochlea + vestibule fused; limited CI outcome |
| Cochlear aplasia | Absent cochlea, present vestibule |
| Mondini malformation | Incomplete partition Type II; 1.5 turns; EVA |
| Enlarged Vestibular Aqueduct (EVA) | >1.5 mm midpoint; commonest radiological finding in SNHL; Pendred |
| Semicircular canal aplasia | Isolated |
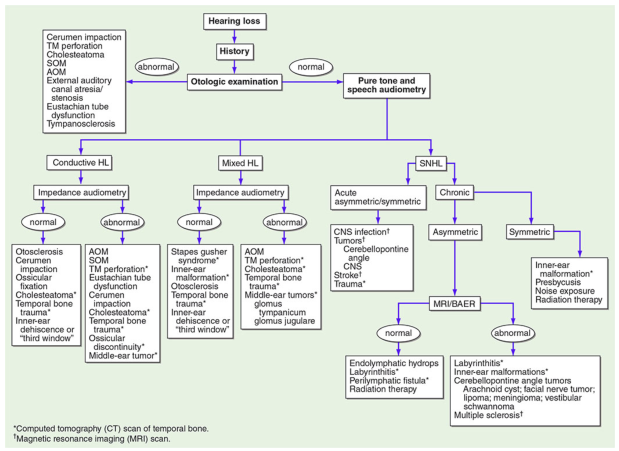
V. LABORATORY / GENETIC WORKUP
| Investigation | Purpose |
|---|---|
| TORCH titres (maternal + neonatal) | Congenital infections |
| Thyroid function tests | Pendred syndrome (euthyroid goitre with positive perchlorate discharge test) |
| Perchlorate discharge test | Pendred syndrome confirmation |
| ECG | Jervell–Lange-Nielsen (prolonged QT) |
| Urinalysis + renal function | Alport syndrome |
| GJB2 / Connexin 26 mutation analysis | Commonest genetic cause; guides prognosis; cochlear implant outcomes excellent |
| GJB6 / Connexin 30 | Large deletion on chromosome 13q |
| SLC26A4 (Pendrin) | Pendred syndrome, non-syndromic EVA |
| Karyotype | Trisomy 21 (Down syndrome — OME + SNHL) |
| MtDNA analysis | Mitochondrial HL (A1555G — aminoglycoside sensitivity) |
| Ophthalmology | Usher syndrome (ERG), Alport (lenticonus) |
| Renal ultrasound | BOR syndrome, Alport |
| VDRL / FTA-ABS | Congenital syphilis |
| CMV PCR (urine/saliva) | Congenital CMV — most common infectious cause today |
MANAGEMENT
Stell & Maran (Head & Neck Surgery, 5th ed); Cummings Otolaryngology (7th ed): Management of deaf mutism must be early, comprehensive, and multidisciplinary. The goal is early auditory rehabilitation followed by speech-language habilitation.
FLOWCHART 3: Management Algorithm
CONFIRMED PROFOUND SNHL / DEAF MUTISM
│
┌─────────┴──────────┐
│ │
Conductive component Pure SNHL
identifiable (no correctable cause)
│ │
▼ │
Surgical correction │
(atresiaplasty, BAHA) │
│ │
└─────────┬──────────┘
│
▼
AGE OF CHILD?
┌─────────┴─────────┐
│ │
< 12 months > 12 months
(optimal window) (still implant
│ if <5 yrs)
▼ │
COCHLEAR IMPLANT ←──────┘
(if profound bilateral SNHL
+ intact cochlear nerve
+ no medical contraindication)
│
▼
POST-IMPLANT REHABILITATION
• Auditory Verbal Therapy (AVT)
• Speech-Language Therapy
• Special education
• Parental counselling
If NOT cochlear implant candidate:
│
▼
HEARING AID FITTING
(if residual hearing >30dB)
+ Speech therapy
+ Special school
+ Sign language (if needed)
I. PREVENTION
| Level | Intervention |
|---|---|
| Primary | Rubella vaccination (MMR); genetic counselling; avoid aminoglycosides in neonates; prevent consanguinity awareness |
| Secondary | Universal Newborn Hearing Screening (UNHS) — detect before 1 month; confirm by 3 months; intervene by 6 months (1-3-6 rule, JCIH 2019) |
| Tertiary | Early cochlear implantation; rehabilitation |
II. HEARING AIDS
Indications: HL >30–40 dB HL; when cochlear implant not feasible or as pre-implant trial
| Type | Use |
|---|---|
| Behind-the-ear (BTE) | Commonest in children; allows binaural fitting |
| In-the-ear (ITE) | Adults; poor fit in growing ears |
| Body-worn | Severe–profound; robust; used in children |
| CROS/BiCROS | Unilateral HL |
| Bone Anchored HA (BAHA) | Conductive/mixed HL; atresia; SNHL with chronic ear discharge |
| Bone-conduction hearing aids | Pre-BAHA implant age (<5 yrs) on headband |
Hearing Aid Selection based on:
- Type and degree of HL (PTA, ASSR)
- Speech discrimination score (SDS — the better the SDS, the better the prognosis with HA)
- Dynamic range of hearing
- Age and cooperation
III. COCHLEAR IMPLANT (CI)
Scott Brown's Otorhinolaryngology, Head & Neck Surgery (8th ed, Gleeson): "Cochlear implantation is the most significant advance in the management of profound sensorineural deafness in children."
A. Candidacy Criteria
| Criteria | Detail |
|---|---|
| Degree of HL | Bilateral profound SNHL (>90 dB HL) in adults; >70 dB HL in children (expanded criteria) |
| Age | Children: ≥12 months (FDA); can be done 6–12 months in some centres; adults: any age |
| Hearing aid trial | Inadequate benefit from optimally fitted HAs (SDS <50% open-set sentences) |
| Cochlear nerve | Must be present (MRI IAM essential) |
| Cochlear anatomy | Cochlea must be patent (CT temporal bone) — Mondini, EVA: can still implant |
| Motivation | Realistic expectations; family commitment to rehabilitation |
| No medical contraindication | General anaesthesia fitness |
B. Contraindications to CI
- Michel aplasia (absent cochlea)
- Cochlear nerve aplasia (absent VIII nerve on MRI)
- Active middle ear infection (relative; should be treated first)
- Central auditory processing disorders (rare)
- Lack of rehabilitative support
C. Components of Cochlear Implant
COCHLEAR IMPLANT SYSTEM
│
├── EXTERNAL
│ ├── Microphone (picks up sound)
│ ├── Sound processor (digitizes, encodes)
│ └── Transmitter coil (sends signal across skin)
│
└── INTERNAL
├── Receiver-stimulator (under skin)
└── Electrode array (inserted into scala tympani
of cochlea — 12–22 electrodes)
→ Directly stimulates spiral ganglion neurons
→ Signal travels via auditory nerve to brain
D. Pre-operative Assessment for CI
- Full audiological workup (PTA, BERA, ASSR, OAE, aided audiogram)
- HRCT temporal bone — cochlear anatomy, patency, facial nerve
- MRI IAM (1.5T or 3T) — cochlear nerve, cochlear morphology
- Speech and language assessment
- Psychological assessment
- Medical fitness (ENT, anaesthesia, paediatrics)
- Vestibular function (rotation tests, VEMPs)
E. Surgical Steps (Simplified)
- Post-auricular incision
- Cortical mastoidectomy
- Posterior tympanotomy (facial recess approach)
- Cochleostomy (adjacent to round window) or round window insertion
- Electrode array insertion into scala tympani
- Receiver-stimulator placed in bony well posterior to mastoid
- Wound closure
- Switch-on: 4–6 weeks post-op; mapping begins
F. Outcomes
- Pre-lingual children implanted <2 years develop near-normal oral language
- Results best when: early implantation, strong family involvement, intensive AVT
- Post-lingual adults: excellent outcomes — speech understanding in quiet ~80–90%
- Pre-lingual adults: poor outcomes — limited by absent auditory memory
IV. BONE ANCHORED HEARING AID (BAHA / BAHS)
Indications in deaf-mute context:
- Conductive/mixed HL with chronic discharge (cannot wear conventional HA)
- Congenital aural atresia (before atresiaplasty or when surgery not feasible)
- Single-sided deafness (SSD)
Principle: Titanium implant in mastoid cortex → osseointegrates → direct bone vibration → cochlea stimulated directly, bypassing middle ear
V. MIDDLE EAR IMPLANTS
- For moderate–severe SNHL where conventional HAs give inadequate benefit
- Vibrant Soundbridge (VSB), Esteem implant
- Transducer attached to ossicular chain or round window
VI. AUDITORY BRAINSTEM IMPLANT (ABI)
- Used when cochlear nerve is absent/non-functional (e.g., NF2, cochlear nerve aplasia)
- Electrode placed on cochlear nucleus in lateral recess of IV ventricle
- Outcomes inferior to CI but better than no device
VII. SPEECH AND LANGUAGE HABILITATION
The most critical aspect post-device fitting.
A. Auditory Verbal Therapy (AVT)
- Goal: teach child to listen and speak; no sign language used
- Intensive, parent-mediated therapy
- Best outcomes when started earliest
B. Auditory Oral Approach
- Listening + lip reading (speech reading) combined
- No sign language
C. Total Communication
- Combines speech, lip reading, sign language, and written language
- Used when oral communication inadequate
D. Sign Language / Cued Speech
- Used when auditory-oral methods fail
- Indian Sign Language (ISL) in India
E. Special Education
- Special school for hearing impaired: structured oral education
- Mainstream school with resource teacher (after CI rehabilitation)
- Individual Educational Plan (IEP)
VIII. TEAM APPROACH — MULTIDISCIPLINARY MANAGEMENT
DEAF-MUTE CHILD
│
├── ENT Surgeon (CI surgery, hearing aid prescription, diagnosis)
├── Audiologist (PTA, OAE, BERA, ASSR, HA fitting, CI programming/mapping)
├── Speech-Language Pathologist (speech therapy, AVT)
├── Paediatrician (infections, jaundice, syndromes)
├── Ophthalmologist (Usher, Waardenburg, Alport)
├── Cardiologist (Jervell–Lange-Nielsen ECG)
├── Nephrologist (Alport)
├── Geneticist (GJB2, SLC26A4 mutation counselling)
├── Psychologist (family counselling, adjustment)
└── Special Educator (IEP, school integration)
RECENT ADVANCES
(Scott Brown 8th ed; Cummings 7th ed; Recent Literature)
| Area | Advance |
|---|---|
| Genetics | Next-generation sequencing (NGS) gene panels detect >100 deafness genes simultaneously; whole exome sequencing |
| Newborn Screening | JCIH 2019: universal OAE + AABR by 1 month; confirm by 3 months; intervene by 6 months |
| CI in <12 months | Implantation as young as 5–6 months now done in specialized centres with excellent outcomes |
| Totally Implantable CI | Totally implantable devices (Esteem, Cochlear's Kanso 2) — no external hardware visible |
| Electric-Acoustic Stimulation (EAS) | Hybrid CI for patients with low-frequency residual hearing — preserves residual acoustic hearing + adds electrical high-frequency stimulation |
| Bilateral CI | Improves spatial hearing and noise performance vs. unilateral; sequential or simultaneous |
| MRI-compatible CI | All modern CI systems MRI-compatible at 1.5T (some at 3T) without magnet removal |
| Gene therapy | Adeno-associated virus (AAV) delivery of OTOF (otoferlin) gene in DFNB9 deafness — Phase I/II trials (2023); early results show restoration of hearing |
| Stem cell therapy | Cochlear hair cell regeneration research — pre-clinical stage; ATOH1 gene stimulates hair cell regeneration in animal models |
| Artificial Intelligence in audiology | AI-based audiogram interpretation, CI fitting algorithms, remote programming (teleaudiology) |
| Remote CI mapping | Teleaudiology — CI programming via internet; COVID-19 accelerated adoption |
| Connexin gene therapy | GJB2 AAV gene therapy pre-clinical studies |
| CMV prevention | Congenital CMV now #1 infectious cause of SNHL; ganciclovir/valganciclovir treatment in CMV-SNHL can stabilize hearing |
PROGNOSIS
| Factor | Favourable | Unfavourable |
|---|---|---|
| Age at intervention | Early (<1 year) | Late (>5 years pre-lingual) |
| Cause | Post-meningitis (before ossification) | Congenital malformation |
| Cochlear anatomy | Normal | Mondini, common cavity |
| Cochlear nerve | Present | Absent/hypoplastic |
| Family support | Excellent | Poor |
| IQ | Normal | Cognitive delay |
| Residual hearing | Present | None |
SUMMARY TABLE — INVESTIGATIONS AT A GLANCE
| Investigation | Purpose | Age Applicability |
|---|---|---|
| OAE (TEOAE/DPOAE) | Cochlear screening | Any age (neonates to adults) |
| AABR / BERA | Neural pathway; threshold | Any age; gold standard neonates |
| ASSR | Frequency-specific thresholds | Infants onwards |
| BOA | Gross behavioural | 0–6 months |
| VRA | Conditioned behavioural | 6 months–2.5 years |
| Play audiometry | Conditioned behavioural | 2.5–5 years |
| PTA | Gold standard threshold | >5 years (cooperative) |
| Speech audiometry | SRT, SDS | >5 years |
| Impedance audiometry | Middle ear; reflexes | Any age |
| HRCT temporal bone | Bony anatomy | Pre-CI workup |
| MRI IAM | Cochlear nerve, inner ear | Pre-CI workup |
| GJB2/connexin 26 | Genetic diagnosis | Any age |
| TORCH, CMV PCR | Infectious cause | Neonates/infants |
| ECG | Jervell–Lange-Nielsen | All syndromic SNHL |
| Thyroid function + perchlorate | Pendred syndrome | All children with SNHL + goitre |
KEY REFERENCES
- Scott Brown's Otorhinolaryngology, Head & Neck Surgery (8th ed, Gleeson) — Vol 3: Otology; Chapters on pediatric deafness, cochlear implant
- Cummings Otolaryngology — Head and Neck Surgery (7th ed, Flint et al.) — Chapter 146: Cochlear Implant; Chapter 145: Pediatric Audiology
- Stell & Maran's Textbook of Head and Neck Surgery and Oncology (5th ed) — Otology section
- Zakir Hussain — Textbook of ENT & Head Neck Surgery — Chapter on Deafness and hearing aids
- Dhingra — Diseases of Ear, Nose & Throat (7th ed) — Chapter 14: Diseases of the Inner Ear; Deafness, Deaf-mutism
- Hazarika — Textbook of Ear, Nose & Throat Diseases — Chapter on Deafness
- Harrison's Principles of Internal Medicine (21st ed, p.1022) — Hearing loss algorithm
- JCIH 2019 Position Statement — Year 2019 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs
- Clinical Evaluation and Etiologic Diagnosis of Hearing Loss — JAMA/Otolaryngology-HNS (retrieved, p.3): Connexin 26 genetics
- Sennaroglu L — Cochlear implantation in inner ear malformations — Cochlear Implants International, 2010
- Omichi R et al. — Gene therapy for hearing loss — Human Molecular Genetics, 2019
- Lenarz T — Cochlear implant — state of the art — GMS Current Topics in Otorhinolaryngology, 2017
This comprehensive answer covers all key domains expected in a 50-mark question: definition, classification, etiology (genetic and acquired), detailed evaluation (history, examination, audiological workup for each age group, radiology, labs), management (prevention, hearing aids, cochlear implants, ABI, rehabilitation, multidisciplinary approach), recent advances, and prognosis — drawing from Scott Brown, Cummings, Stell & Maran, Zakir Hussain, Dhingra, Hazarika, and current literature.
This is a shared conversation. Sign in to Orris to start your own chat.