Acute leukemias pathology MBBS standard text book
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have all the information needed to write a comprehensive MBBS-level pathology response on acute leukemias. Let me compile the complete answer.
Acute Leukemias - Pathology (MBBS Standard)
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease
Definition and Overview
Acute leukemias are aggressive neoplasms of immature hematopoietic precursor cells (blasts) that undergo maturation arrest, accumulate in the bone marrow, and suppress normal hematopoiesis. There are two major categories:
| Feature | ALL (Acute Lymphoblastic Leukemia/Lymphoma) | AML (Acute Myeloid Leukemia) |
|---|---|---|
| Cell of origin | Precursor B or T lymphoblasts | Myeloid progenitors |
| Peak age | Children (~3 yrs for B-ALL; adolescents for T-ALL) | Adults (incidence rises with age, peaks >60 yrs) |
| Annual US cases | ~2,500 | ~13,000 |
| Blast marker | TdT+, MPO- | MPO+, TdT- |
| Pathognomic finding | None | Auer rods |
PART 1: Acute Lymphoblastic Leukemia / Lymphoma (ALL)
Subtypes
- B-ALL: ~85% of all ALL; arises from pre-B cells; typical childhood leukemia
- T-ALL: ~15%; arises from thymic T-cell precursors; adolescent males; often presents as mediastinal mass
Pathogenesis
Mutations target "master" transcription factors required for lymphoid development, causing maturation arrest and stem-cell-like self-renewal:
- B-ALL driver genes: PAX5, TCF3, ETV6, RUNX1, BCR::ABL1, KMT2A, PBX1
- T-ALL: predominantly NOTCH1 mutations (50-70%), which is essential for T-cell development
Important chromosomal aberrations in B-ALL:
| Aberration | Frequency | Significance |
|---|---|---|
| t(12;21) - ETV6::RUNX1 | 25% | Favorable prognosis |
| Hyperdiploidy (>50 chr) | Common | Favorable prognosis |
| Hypodiploidy | Less common | Poor prognosis |
| t(9;22) - BCR::ABL1 (Philadelphia) | Variable | Targetable with TKI; BCR-ABL is 190 kDa in ALL vs 210 kDa in CML |
| KMT2A rearrangements | Infantile ALL | Very poor; associated with CNS disease |
Morphology
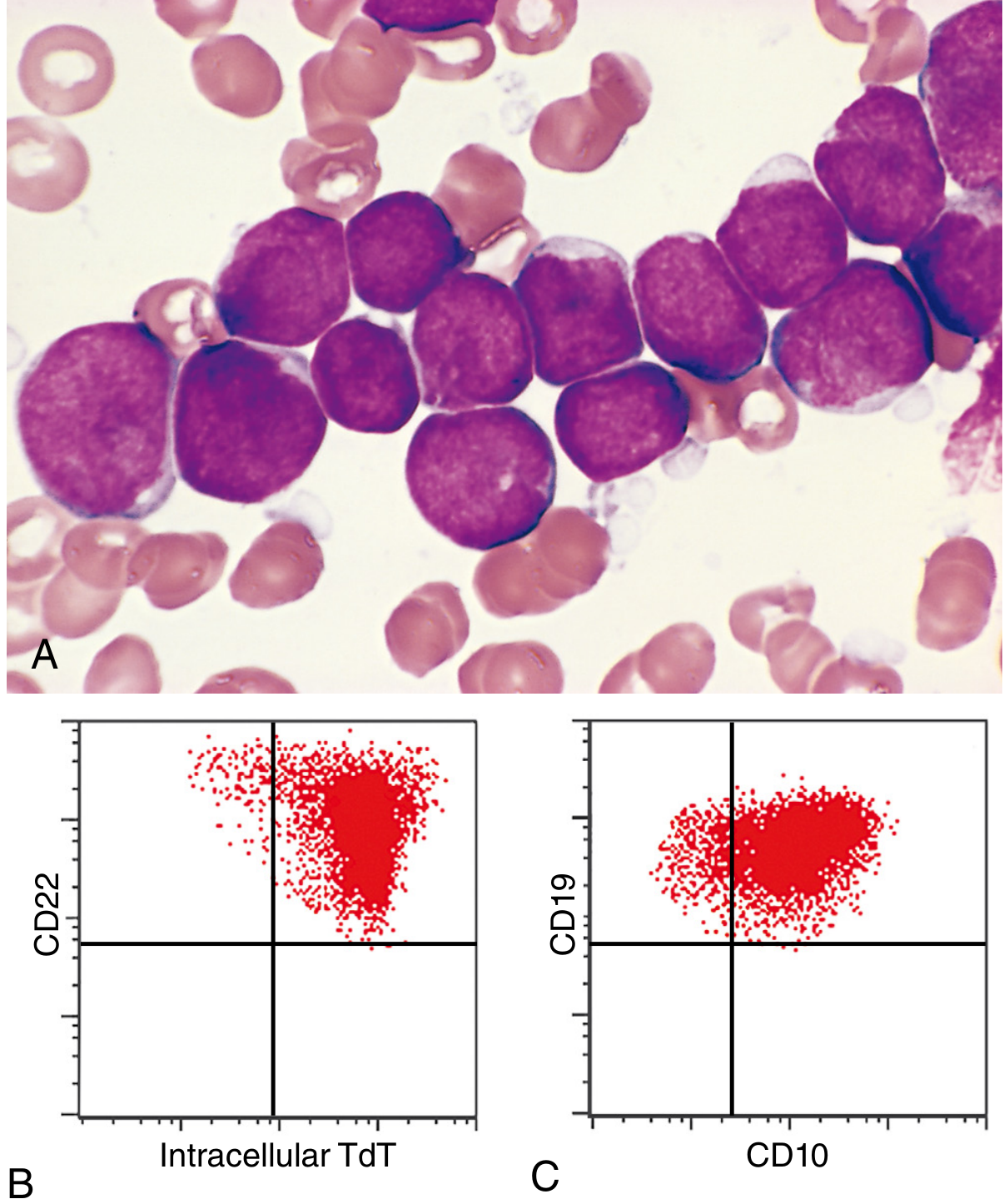
The bone marrow is hypercellular, packed with lymphoblasts replacing normal elements.
Lymphoblast morphology:
- Scant basophilic cytoplasm (agranular)
- Nuclei somewhat larger than small lymphocytes
- Delicate, finely stippled nuclear chromatin
- Small but sharply demarcated nucleoli
- Nuclear membrane often deeply subdivided (convoluted appearance)
- High mitotic rate
- Interspersed macrophages create a "starry sky" appearance (in rapidly growing tumors)
In T-ALL: mediastinal thymic masses occur in 50-70%, along with lymphadenopathy and splenomegaly.
Immunophenotype
TdT (Terminal Deoxynucleotidyl Transferase) - positive in >95% of ALL; this specialized DNA polymerase is expressed only in pre-B and pre-T lymphoblasts - the single most important distinguishing marker from AML.
| Subtype | Markers |
|---|---|
| B-ALL (early) | TdT+, CD19+, PAX5+, CD10- |
| B-ALL (late pre-B) | TdT+, CD10+, CD19+, CD20+, cytoplasmic IgM |
| T-ALL (immature) | TdT+, CD1, CD2, CD5, CD7; CD3-/CD4-/CD8- |
| T-ALL (late) | CD3+, CD4+, CD8+ |
Key histochemical contrast with AML:
- ALL: MPO-negative, PAS-positive cytoplasmic material
- AML: MPO-positive, Auer rods
Clinical Features
- Abrupt, stormy onset within days to weeks
- Anemia: fatigue, pallor
- Neutropenia: fever, recurrent infections
- Thrombocytopenia: bleeding, petechiae, ecchymoses
- Mass effects (more prominent than in AML):
- Bone pain from marrow expansion/periosteal infiltration
- Generalized lymphadenopathy, hepatosplenomegaly
- Testicular enlargement
- Mediastinal mass (T-ALL) causing vascular/airway compression
- CNS spread: headache, vomiting, cranial nerve palsies (more common than AML)
Prognosis - ALL
Children: ~95% complete remission; 75-85% cured (one of oncology's greatest successes)
Poor prognostic factors:
- Age <2 years (KMT2A rearrangements, CNS involvement)
- Adolescence or adulthood
- Blast count >100,000/µL (high tumor burden)
Favorable prognostic factors:
- Age 2-10 years
- Low WBC
- Hyperdiploidy
- Trisomy of chromosomes 4, 7, 10
- t(12;21) - ETV6::RUNX1
Modern therapies: CAR-T cells targeting CD19 have produced dramatic responses in B-ALL; BCR-ABL kinase inhibitors (imatinib, dasatinib) are key for Ph+ ALL.
PART 2: Acute Myeloid Leukemia (AML)
Definition
AML is a tumor of hematopoietic progenitors caused by acquired oncogenic mutations that impede myeloid differentiation, leading to accumulation of immature myeloid blasts. Diagnosis requires ≥20% blasts in the bone marrow (exception: if a defining genetic aberration is present, diagnosis can be made with fewer blasts).
WHO Classification (Two Major Categories)
I. AML with Specific Genetic Aberrations:
| Subtype | Prognosis | Morphology/Notes |
|---|---|---|
| t(8;21) - RUNX1::RUNX1T1 | Favorable | Full myelocytic maturation; Auer rods easily found |
| inv(16) - CBFB::MYH11 | Favorable | Mixed myelocytic + monocytic; abnormal eosinophilic precursors |
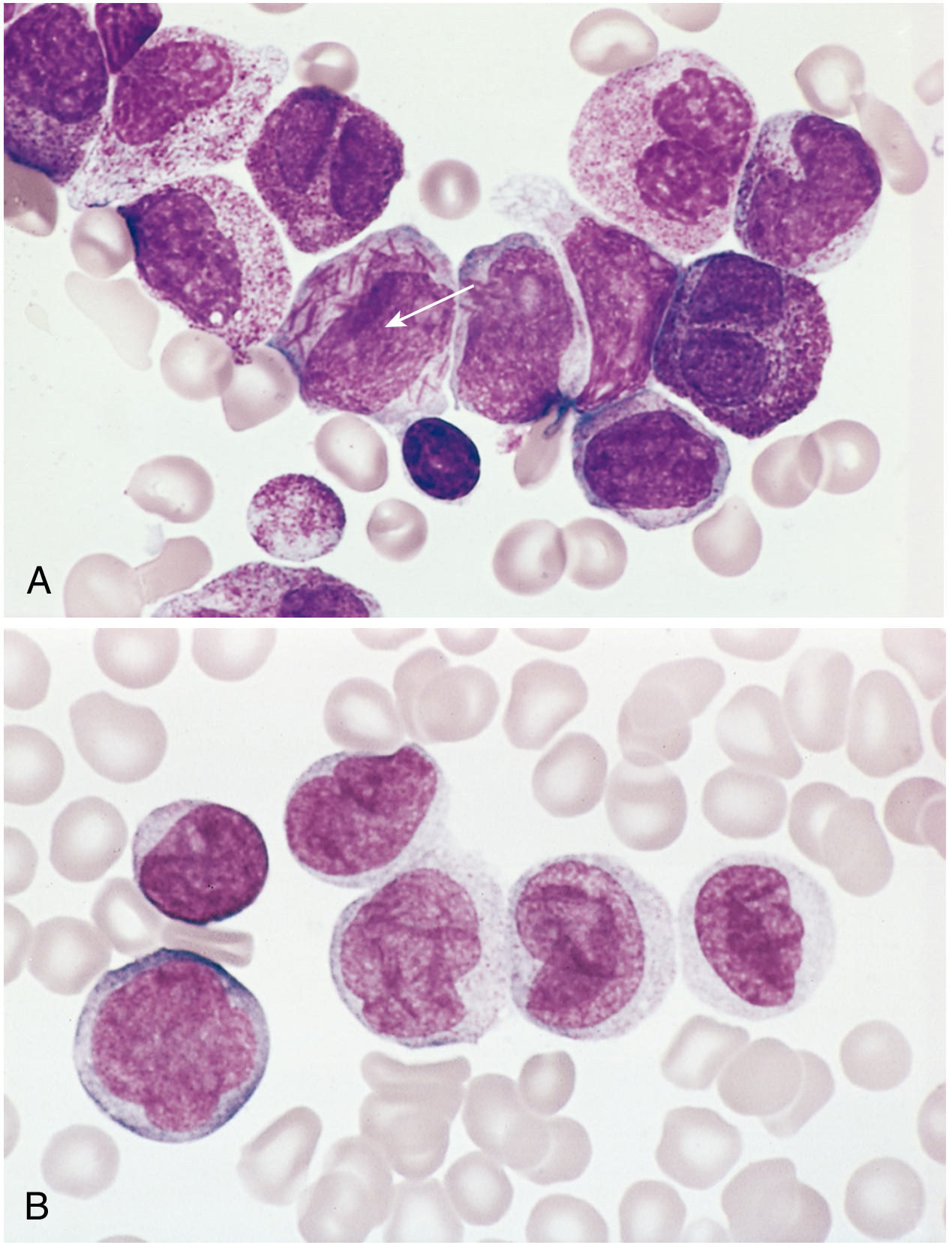
| t(15;17) - PML::RARA (Acute Promyelocytic Leukemia, APL) | Very favorable | Numerous Auer rods, often in bundles ("faggot cells"); high DIC risk |
| t(11q23) - KMT2A rearrangement | Poor | Monocytic differentiation |
| Mutated NPM1 | Favorable | Detected by sequencing |
| Myelodysplasia-related genetics | Poor | Del 5q/7q; SRSF2, ASXL1, EZH2 mutations |
II. AML Defined by Differentiation:
Minimally differentiated, without maturation, with maturation, myelomonocytic, monocytic, erythroid, megakaryocytic (intermediate prognosis for all)
Pathogenesis - Four Categories of Driver Mutations
- Transcription factor mutations - block myeloid differentiation
- t(8;21): disrupts RUNX1/CBFB complex
- t(15;17): PML-RARα fusion blocks retinoic acid-driven maturation
- Signaling mutations - activate proliferation/survival pathways
- FLT3 activating mutations (co-operate with PML-RARα)
- RAS mutations
- Epigenome mutations - alter DNA methylation or chromatin organization
- IDH1/IDH2 mutations - produce oncometabolite 2-hydroxyglutarate
- Cohesin complex mutations
- TP53 mutations - complex karyotype, erythroid differentiation, very poor prognosis
Morphology
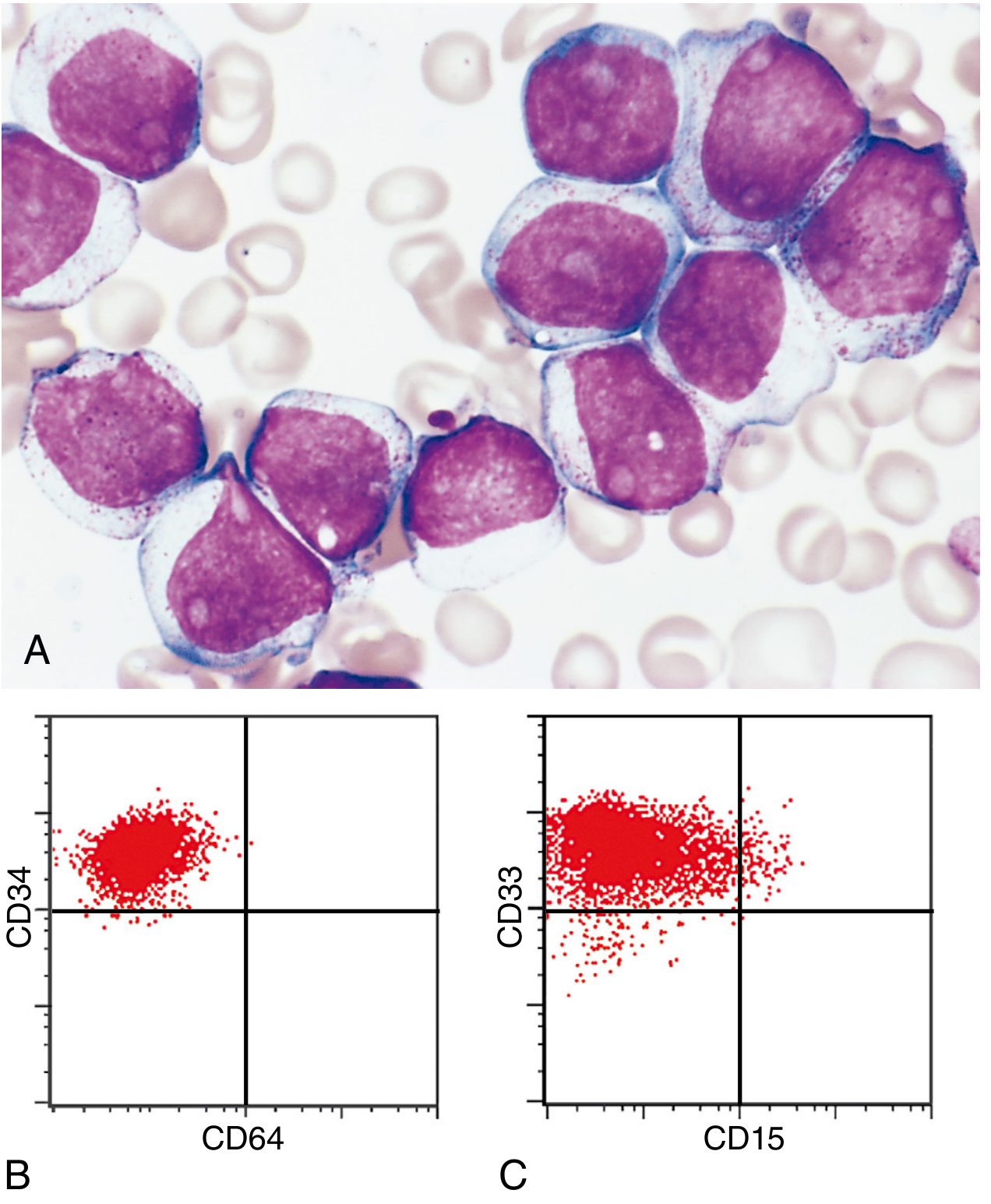
Myeloblast morphology:
- Delicate nuclear chromatin
- 2-4 prominent nucleoli (more than lymphoblasts)
- Voluminous cytoplasm (more than lymphoblasts)
- Fine, peroxidase-positive azurophilic granules
- Auer rods - needle-like azurophilic granules (pathognomonic); most numerous in APL with t(15;17)
- Monoblasts: folded/lobulated nuclei, no Auer rods, nonspecific esterase-positive
- Aleukemic leukemia: occasionally blasts absent from blood - bone marrow biopsy is essential in any pancytopenic patient
Immunophenotype
Myeloid markers (used to distinguish from ALL): CD13, CD33, CD34, CD64, CD15, MPO (myeloperoxidase), nonspecific esterase (for monocytic subtypes).
Cytogenetics
- Chromosomal aberrations detected in 50-70% by standard methods; ~90% by high-resolution banding
- Young adults (de novo): t(8;21), inv(16), t(15;17) - generally favorable
- Post-chemo/radiation: deletions of chr 5 and 7, TP53 mutations - poor
- Post-topoisomerase II inhibitor therapy: KMT2A rearrangements (11q23)
- Elderly: often have mutations shared with clonal hematopoiesis (TET2, DNMT3A) and MDS (SRSF2, etc.)
Clinical Features
Presents within weeks of symptom onset:
- Fatigue (anemia)
- Fever (neutropenia → opportunistic infections: fungi, Pseudomonas, commensals)
- Bleeding (thrombocytopenia): petechiae, ecchymoses, serosal/mucosal hemorrhages; worsened by DIC especially in APL
- Extramedullary spread: less prominent than ALL, but monocytic AML frequently infiltrates skin (leukemia cutis) and gingiva
- CNS spread: less common than ALL
- Myeloid sarcoma: localized soft tissue AML mass (inevitably becomes systemic without treatment)
Prognosis - AML
- ~60% achieve complete remission with chemotherapy
- Only 15-30% disease-free at 5 years overall
- APL t(15;17): >90% curable with all-trans retinoic acid (ATRA) + arsenic trioxide - best prognosis of any AML subtype
- t(8;21) and inv(16): relatively good prognosis
- Older adults, post-MDS, post-genotoxic AML: very poor; treated with HSC transplantation when possible
- Newer targeted therapies: IDH inhibitors (IDH-mutated AML), FLT3 inhibitors (FLT3-mutated AML), BCL2 inhibitors + hypomethylating agents (elderly patients)
Key Concepts Summary
| Feature | ALL | AML |
|---|---|---|
| Cell | Lymphoblast (pre-B or pre-T) | Myeloblast |
| TdT | Positive (>95%) | Negative |
| MPO | Negative | Positive |
| Auer rods | Absent | Present (pathognomonic) |
| PAS stain | Positive | Negative |
| NSE | Negative | Positive (monocytic AML) |
| Hallmark translocation | t(9;22), t(12;21), KMT2A | t(15;17), t(8;21), inv(16) |
| CNS spread | Common | Less common |
| Prognosis (overall) | Good in children (75-85% cure) | Poor (15-30% 5-yr survival) |
| Best subtype outcome | Ph+ ALL with TKI | APL (ATRA + arsenic - >90% cure) |
Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 556-580
This is a shared conversation. Sign in to Orris to start your own chat.