Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive, well-structured answer. Here is the full explanation:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Definition & Berlin Criteria
ARDS is defined by the Berlin criteria: bilateral pulmonary opacities on chest radiograph/CT not fully explained by effusions or collapse; acute onset within 1 week of a known clinical insult; respiratory failure not fully explained by cardiac failure or fluid overload; and hypoxemia stratified by P/F ratio (PaO₂/FiO₂):
| Severity | P/F Ratio |
|---|---|
| Mild | 200–300 mmHg |
| Moderate | 100–200 mmHg |
| Severe | < 100 mmHg |
— Harrison's Principles of Internal Medicine 22E
Precipitating Causes
ARDS arises from direct or indirect lung injury:
| Direct | Indirect |
|---|---|
| Pneumonia | Sepsis |
| Aspiration of gastric contents | Severe trauma / burns |
| Pulmonary contusion | Pancreatitis |
| Near-drowning / toxic inhalation | Multiple transfusions |
| Drug overdose / cardiopulmonary bypass |
Three Phases of ARDS

Figure: Time course of ARDS. The exudative phase (days 0–7) features alveolar edema and hyaline membrane formation; the proliferative phase (days 7–21) features interstitial inflammation; the fibrotic phase (day 21+) features progressive fibrosis and bullae. — Harrison's
Phase 1: Exudative (Days 0–7)
This is the central pathological event in ARDS.
Step 1 — Alveolar-Capillary Barrier Injury
A triggering insult (infectious, ischemic, toxic, or traumatic) initiates excessive and dysregulated inflammation. The two cellular targets are:
- Capillary endothelial cells — loss of the vascular barrier
- Type I pneumocytes (alveolar epithelial cells) — loss of the air-space barrier
Normally, tight junctions between these cells prevent fluid and macromolecules from entering the airspace. In ARDS, these junctions are disrupted by neutrophil mediators, reactive oxygen species (ROS), and proteases, causing paracellular leak. — Murray & Nadel's Textbook of Respiratory Medicine
Step 2 — Neutrophil-Mediated Injury (Central Mechanism)
Microbial products or systemic inflammatory signals are recognized by resident alveolar macrophages, which secrete chemoattractants (IL-8, TNF-α, CCL/CXCL chemokines) that recruit massive numbers of neutrophils into the alveolar and interstitial spaces.
Activated neutrophils then release cytotoxic mediators into the extracellular space:
| Mediator | Effect |
|---|---|
| Neutrophil elastase | Degrades extracellular matrix; disrupts intercellular junctions; induces endothelial and epithelial cell death; levels in BAL correlate with injury severity |
| Matrix metalloproteinases (MMPs) | Degrade junctional proteins in epithelia and endothelia; released by both neutrophils and macrophages |
| Reactive oxygen/nitrogen species (ROS/RNS) | Cause epithelial and endothelial cell death; disrupt tight junctions; overwhelm endogenous antioxidant defenses; potentiate proteinase-induced injury by inactivating antiproteinases |
| Neutrophil extracellular traps (NETs) | Directly injure epithelial and endothelial cells |
| Cationic antimicrobial peptides (defensins) | Contribute to membrane disruption |
Importantly, ARDS can occur even in neutropenic patients, meaning neutrophils, while central, are not the sole mechanism. — Murray & Nadel's
Step 3 — Macrophage and Platelet Amplification
After the initial neutrophilic wave, recruited monocytes differentiate into macrophages and further amplify injury by releasing:
- TNF-α, interferon-β, TRAIL (apoptosis-inducing ligand)
- ROS/RNS and MMPs
- Vascular endothelial growth factor (VEGF)
Platelet-neutrophil complexes (via P-selectin / PSGL-1 interactions) are increasingly recognized as critical drivers of increased vascular permeability. Free hemoglobin from RBCs and angiopoietin-2 released from the pulmonary endothelium further amplify endothelial injury. — Murray & Nadel's; Harrison's
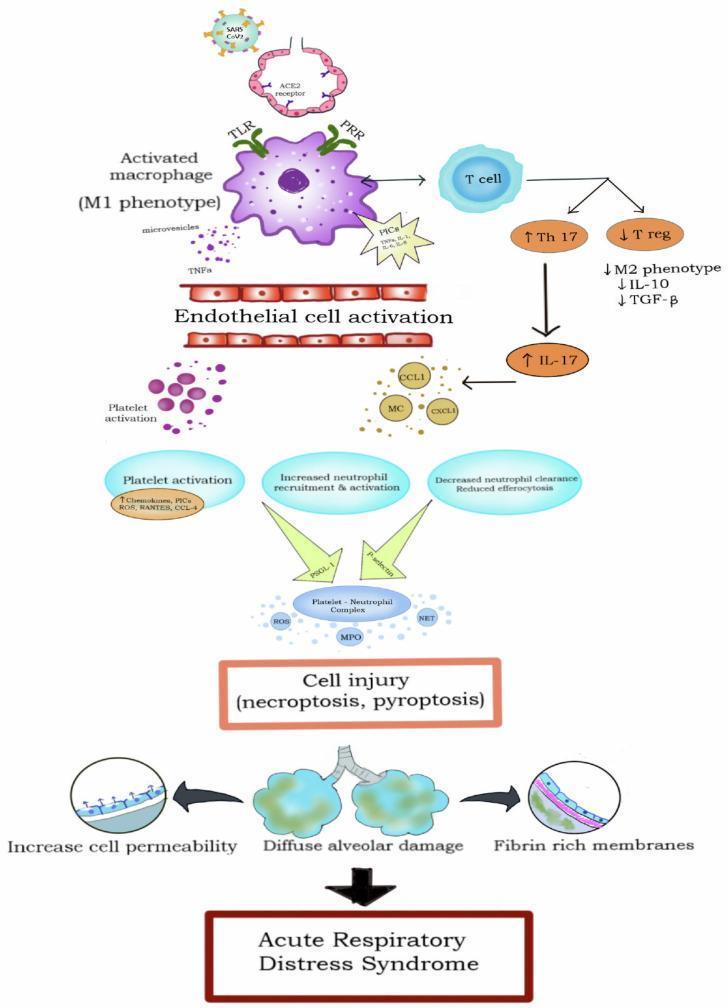
Step 4 — Protein-Rich Alveolar Flooding
The disrupted barrier allows protein-rich edema fluid to flood the interstitium and alveolar spaces. This fluid contains:
- Plasma proteins (albumin, fibrinogen)
- Inflammatory cells and debris
- Inactivated surfactant
Surfactant dysfunction is a key consequence: phospholipase A₂ (elevated in pancreatitis-associated ARDS) degrades surfactant directly, and the protein-rich edema inactivates it functionally. Loss of surfactant raises alveolar surface tension, causing alveolar collapse and further atelectasis. — Murray & Nadel's; Fishman's Pulmonary Diseases
Step 5 — Hyaline Membrane Formation
Fibrin-rich proteinaceous exudate lines the denuded alveolar walls, forming the hallmark hyaline membranes seen on histology (diffuse alveolar damage pattern). This represents the combination of fibrin deposition, cellular debris, and precipitated plasma proteins. — Harrison's
Step 6 — Physiologic Consequences of the Exudative Phase
| Mechanism | Consequence |
|---|---|
| Alveolar flooding and collapse (dependent zones) | Intrapulmonary shunting → refractory hypoxemia |
| Reduced lung compliance (ΔV/ΔP) | Increased work of breathing → dyspnea, respiratory failure |
| Microvascular occlusion | Increased dead space + pulmonary hypertension → hypercapnia |
| Surfactant loss | Alveolar instability, further collapse |
Because edema is gravity-dependent, the dependent lung zones are predominantly consolidated or atelectatic ("sponge lung"), while non-dependent zones may be relatively preserved — the basis of the CT heterogeneity pattern and the rationale for prone positioning. — Goldman-Cecil Medicine; Fishman's
Phase 2: Proliferative (Days 7–21)
In those who survive the exudative phase, a reparative process begins:
- Type II pneumocytes migrate, proliferate, and differentiate to repopulate denuded basement membranes, restoring the epithelial barrier
- Alveolar fluid clearance resumes via active sodium transport across the epithelium (Na⁺/K⁺-ATPase and apical sodium channels on type II cells)
- Interstitial inflammation becomes prominent
- Early fibrotic changes (fibroblast proliferation, collagen deposition) begin in some patients
Many patients recover during this phase. Those who do not may progress. — Harrison's; Fishman's
Phase 3: Fibrotic (Day 21+)
A minority of patients develop a fibrotic phase characterized by:
- Extensive parenchymal fibrosis
- Bullae formation
- Severe, persistent hypoxemia and decreased compliance
- High mortality
The transition to fibrosis represents failure of orderly repair: dysregulated fibroblast activation, ongoing TGF-β signaling, and impaired resolution of inflammation. — Harrison's
Coagulation Dysregulation
ARDS involves simultaneous pro-coagulant and fibrinolytic dysfunction in the alveolar compartment:
- Tissue factor-driven coagulation activation
- Deposition of fibrin in alveolar spaces (contributing to hyaline membranes and microvascular thrombosis)
- Impaired local fibrinolysis
These coagulation disturbances propagate vascular occlusion, dead-space physiology, and pulmonary hypertension. — Murray & Nadel's
Ventilator-Induced Lung Injury (VILI) — a Second-Hit Mechanism
Mechanical ventilation, while life-saving, can worsen ARDS via:
- Volutrauma/barotrauma — overdistension of less-injured lung units with high tidal volumes or pressures disrupts the epithelial-endothelial barrier further
- Atelectrauma — cyclic opening and closing of alveoli at low PEEP shears the airway epithelium
- Biotrauma — mechanical stretch releases inflammatory cytokines (IL-6, IL-8, TNF-α) from alveolar macrophages and epithelial cells into both the lungs and systemic circulation, driving multi-organ failure
This is why lung-protective ventilation (6 mL/kg predicted body weight, plateau pressure ≤30 cmH₂O, PEEP titration, PaO₂ 55–80 mmHg) is the cornerstone of ARDS management. — Fishman's; Goldman-Cecil
Summary: Key Mechanistic Steps
Precipitating insult (direct or indirect)
↓
Alveolar macrophage activation → cytokine/chemokine release
↓
Massive neutrophil (and monocyte) recruitment to alveoli
↓
Release of elastase, MMPs, ROS, NETs → barrier disruption
↓
Loss of tight junctions in endothelium + type I pneumocytes
↓
Protein-rich edema floods alveoli + surfactant inactivation
↓
Hyaline membrane formation (diffuse alveolar damage)
↓
Intrapulmonary shunting + dead-space physiology
↓
Refractory hypoxemia + hypercapnia + pulmonary hypertension
↓
Repair (type II pneumocyte proliferation) OR fibrosis
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Harrison's Principles of Internal Medicine 22E; Fishman's Pulmonary Diseases and Disorders; Goldman-Cecil Medicine; Sabiston Textbook of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.