Explain the pathogenesis of hypertension in flowchart
Detailed medical flowchart showing the pathogenesis of hypertension. Clean white background, professional academic style with boxes and arrows. Layout (top to bottom): TOP ROW - Risk Factors boxes side by side: [Genetic factors (RAAS gene polymorphisms, ENaC mutations, WNK kinase mutations)] [Environmental factors (High salt intake, Obesity, Stress, Smoking, Physical inactivity)] Both arrows converge downward to TWO parallel pathways: LEFT PATHWAY - "RENAL MECHANISM": [Reduced renal Na+ excretion] → [↑ Extracellular fluid volume] → [↑ Blood volume] → [↑ Venous return / Cardiac preload] → [↑ Cardiac Output] RIGHT PATHWAY - "NEUROHUMORAL MECHANISM": [RAAS Activation] → [↑ Renin → Angiotensin I → Angiotensin II → Aldosterone] splits into two sub-branches: Left sub-branch: [Aldosterone → Na+ & H2O retention → ↑ Blood volume → ↑ Cardiac Output] Right sub-branch: [Angiotensin II → Vasoconstriction + SNS activation] [SNS Activation] box (fed by Ang II, obesity, stress, OSA/chemoreflex, baroreflex dysfunction) → branches to: [↑ Norepinephrine → Vasoconstriction] [↑ Heart rate & contractility → ↑ Cardiac Output] [Renal Na+ retention → salt sensitivity] [Endothelial Dysfunction] box → [↓ Nitric Oxide → impaired vasodilation → ↑ Vasoconstriction] All pathway endpoints (↑ Cardiac Output and ↑ Peripheral Vascular Resistance) converge into: [HYPERTENSION: BP = Cardiac Output × Total Peripheral Resistance] (large prominent box) BOTTOM ROW - End-organ damage boxes: [Hypertensive Nephrosclerosis] [Left Ventricular Hypertrophy] [Stroke / Cerebral Hemorrhage] [Accelerated Atherosclerosis] [Arteriolosclerosis (hyaline & hyperplastic)] Use colored boxes: blue for risk factors, orange for mechanisms, red for hypertension result, grey for end-organ damage. Clear directional arrows throughout.
Pathogenesis of Hypertension - Explained
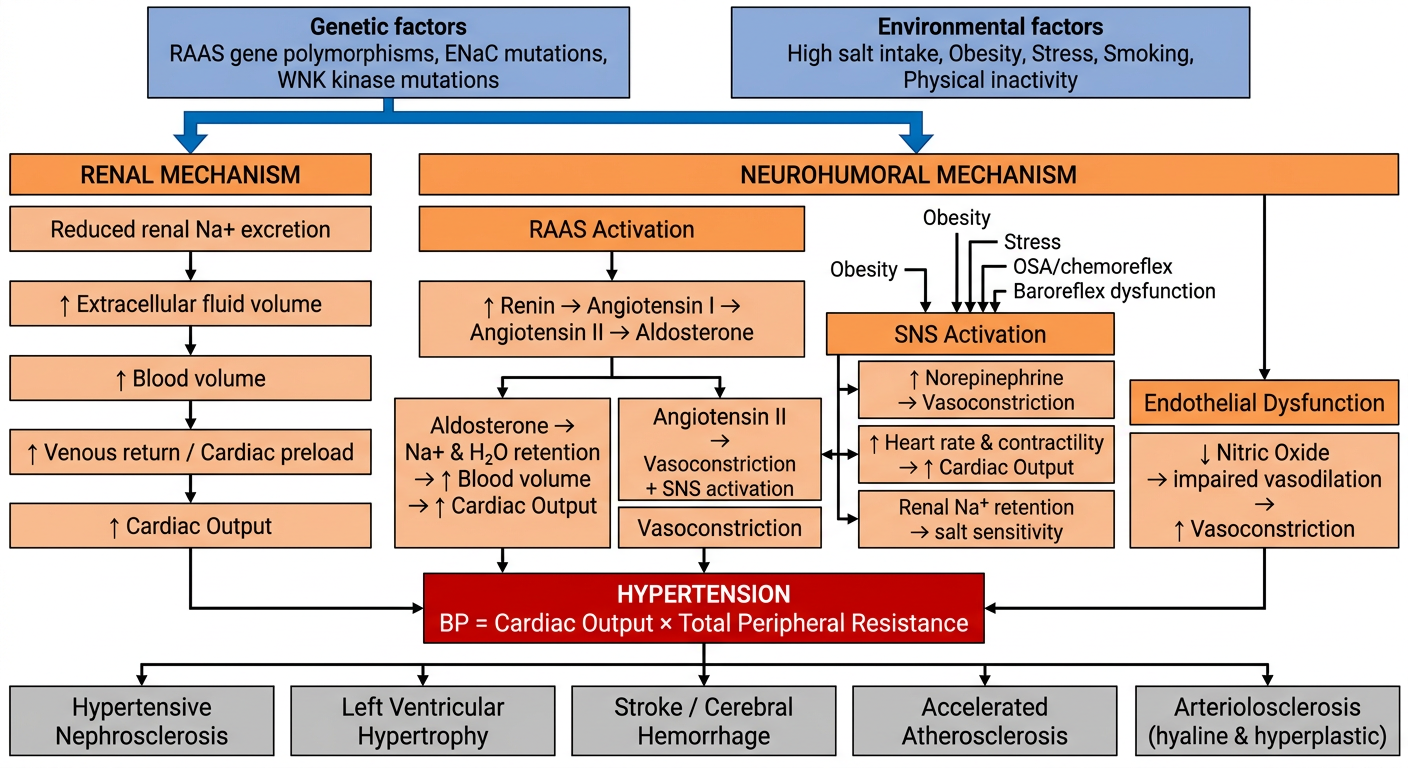
1. Starting Point: Risk Factors
- Polymorphisms in the angiotensinogen gene and AT1 receptors alter RAAS responsiveness
- Monogenic causes (e.g., Liddle syndrome - gain-of-function ENaC mutations; Gordon syndrome - WNK kinase mutations causing increased NCC activity) all share one common feature: defective renal sodium handling
- GWAS studies have identified >100 risk loci, though each contributes only ~0.5-1 mmHg
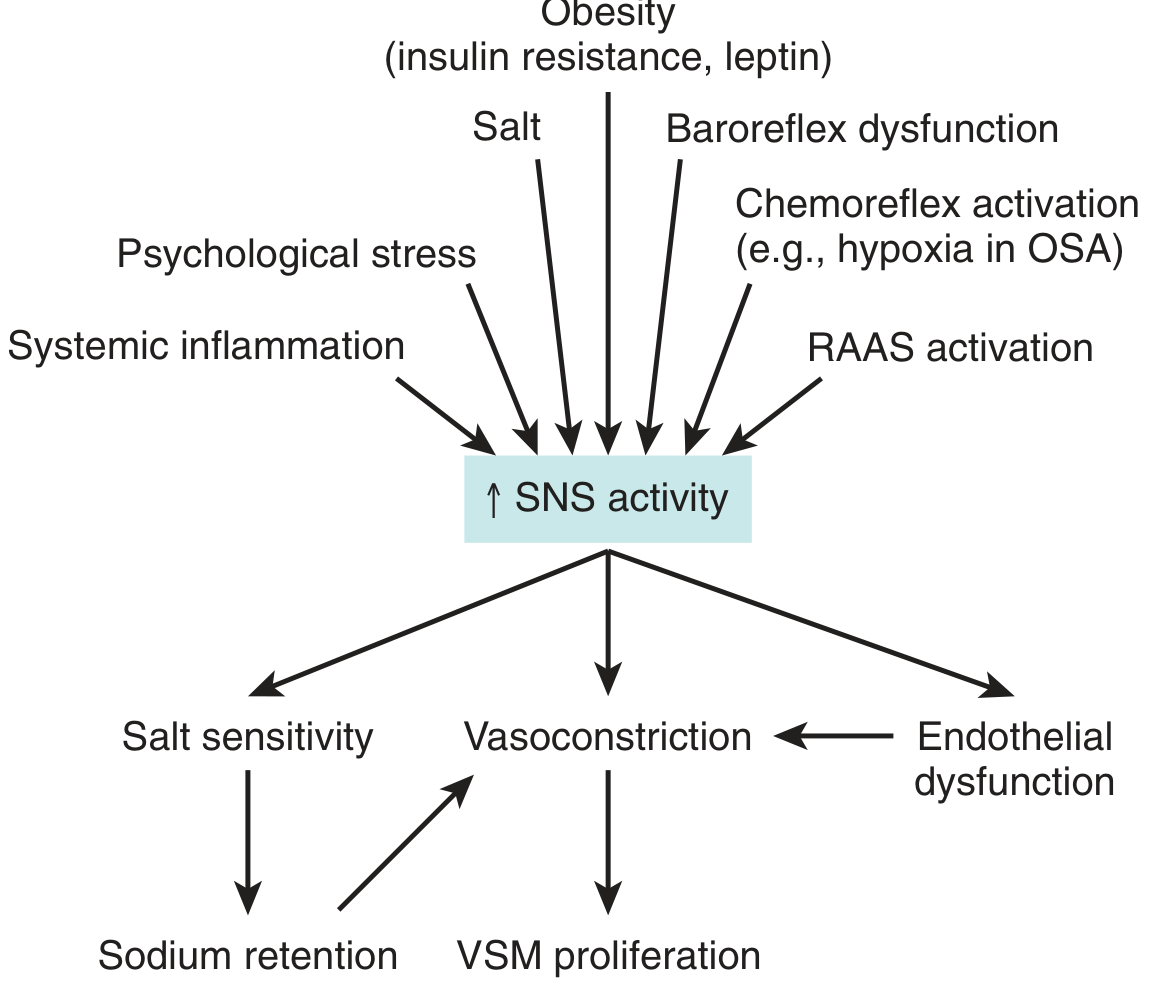
- High salt intake, obesity, stress, smoking, physical inactivity
- Obesity is particularly relevant - it drives both SNS hyperactivity and RAAS activation via insulin resistance and excess leptin
2. Two Core Mechanisms Driving BP Up
"Reduced renal sodium excretion in the presence of normal arterial pressure is probably a key pathogenic feature - this is a common etiologic factor in most forms of hypertension." - Robbins & Kumar Basic Pathology
- Impaired Na+ excretion → ↑ ECF volume → ↑ blood volume → ↑ venous return → ↑ Cardiac Output (CO)
- The kidney eventually excretes more Na+ at the new higher BP (pressure natriuresis), establishing a new steady state at the cost of sustained hypertension
- Low renal perfusion / low Na+ delivery → juxtaglomerular cells release Renin
- Renin cleaves angiotensinogen → Angiotensin I
- ACE converts Ang I → Angiotensin II
- Ang II acts via AT1 receptors to:
- Cause direct vasoconstriction (↑ SVR)
- Stimulate aldosterone release from adrenal cortex → Na+/H2O retention → ↑ blood volume → ↑ CO
- Promote SNS activation
- Drive endothelial dysfunction and vascular remodeling
- ↑ Norepinephrine → vasoconstriction → ↑ Total Peripheral Resistance (TPR)
- ↑ Heart rate & contractility → ↑ CO
- Renal Na+ retention → salt sensitivity
- Vascular smooth muscle (VSM) proliferation → structural thickening → permanent ↑ TPR
- ↓ Nitric oxide (NO) production → impaired vasodilation → ↑ vasoconstriction
- Enhanced by Ang II, oxidative stress, and aldosterone
3. Final Common Pathway
BP = Cardiac Output × Total Peripheral Resistance
- ↑ CO (from volume expansion and SNS-driven cardiac effects)
- ↑ TPR (from vasoconstriction, endothelial dysfunction, and structural vascular remodeling)
4. End-Organ Consequences
| Organ | Consequence |
|---|---|
| Kidney | Hypertensive nephrosclerosis (hyaline arteriolosclerosis → glomerulosclerosis) |
| Heart | Left ventricular hypertrophy, heart failure, coronary artery disease |
| Brain | Stroke, cerebral hemorrhage, lacunar infarcts |
| Vasculature | Accelerated atherosclerosis, aortic dissection |
| Small vessels | Hyaline arteriolosclerosis (benign HTN) or hyperplastic "onion-skin" arteriolosclerosis (malignant HTN) |
Key Diagram from Brenner & Rector's The Kidney