Primary amenorrhea from gynecology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from Berek & Novak's Gynecology plus the full diagnostic flowchart (two parts). Let me compile the complete answer.
Primary Amenorrhea
Source: Berek & Novak's Gynecology, Chapter 34 – Amenorrhea
Definition
Primary amenorrhea is defined as:
- Absence of menses by age 13 when there is no visible development of secondary sexual characteristics, OR
- Absence of menses by age 15 in the presence of normal secondary sexual characteristics
These age cutoffs were revised downward by 1 year to remain at 2 standard deviations above the mean age of developing secondary sexual characteristics and menarche. Failure to begin breast development by age 13 always warrants investigation.
The physiologic prerequisites for menstruation are: pulsatile GnRH from the hypothalamus → LH and FSH from the pituitary → follicular development and ovulation → ovarian estrogen and progesterone production → endometrial cycling. Failure at any level (hypothalamus, pituitary, ovary, uterus, outflow tract) prevents menstruation.
Diagnostic Flowchart (Fig. 34-1)
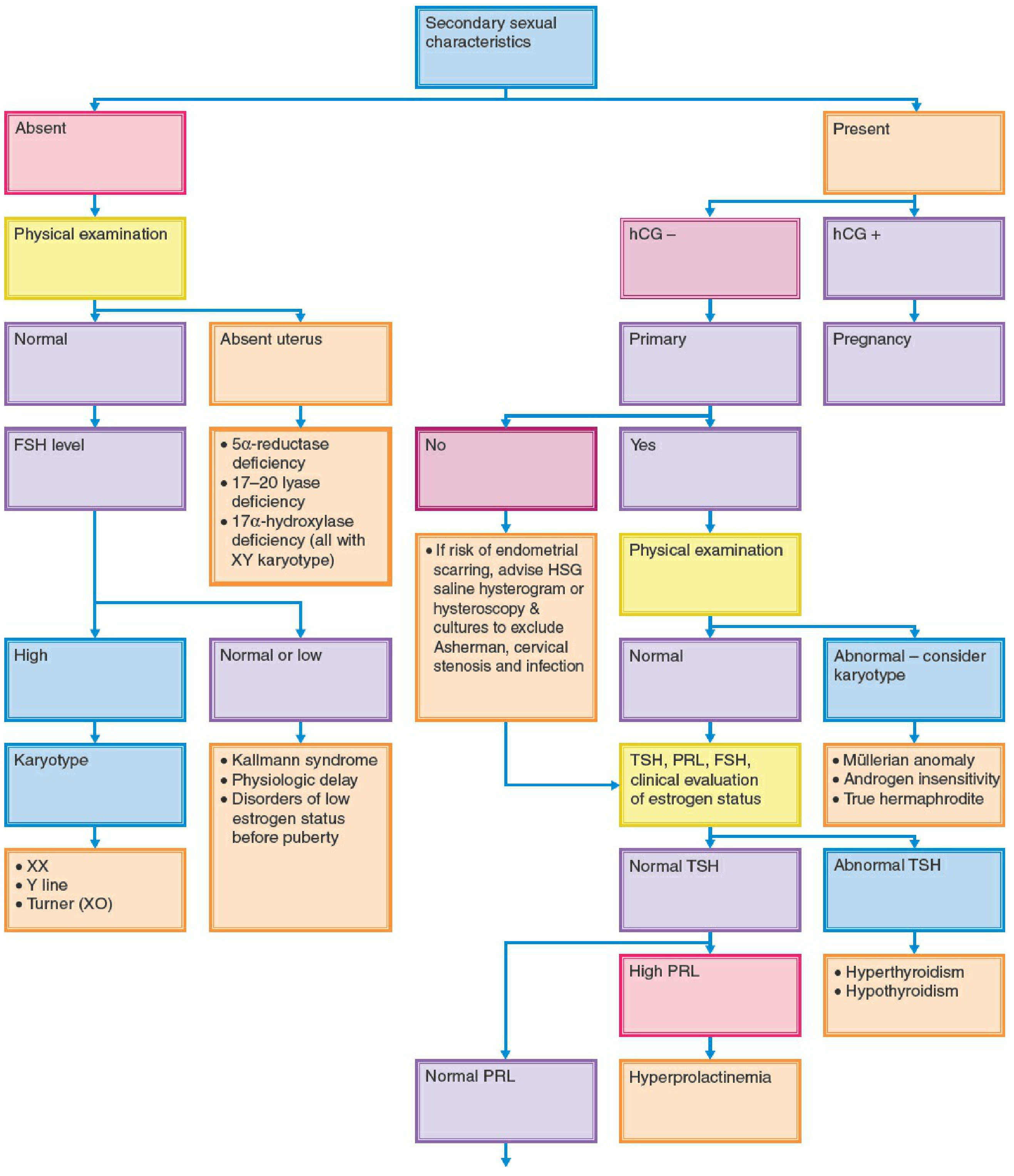
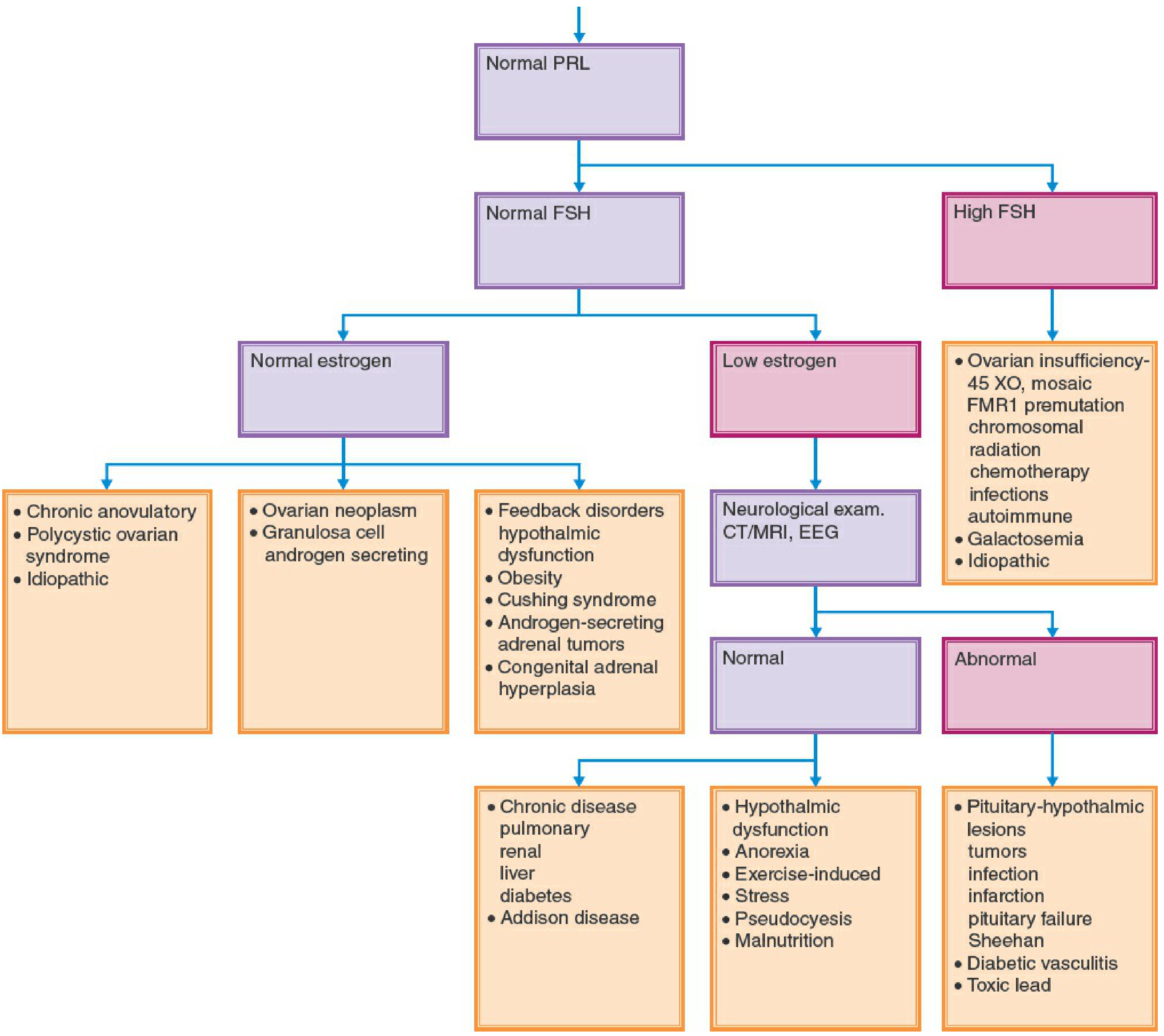
Classification Framework
Primary amenorrhea is best categorized by asking two questions:
- Are secondary sexual characteristics (breasts) present?
- Is pelvic anatomy normal?
This gives three clinical scenarios:
| Scenario | Key Associations |
|---|---|
| No secondary sexual characteristics | Hypergonadotropic or hypogonadotropic hypogonadism |
| Secondary characteristics present + abnormal pelvic anatomy | Outflow/Müllerian anomalies |
| Secondary characteristics present + normal pelvic anatomy | PCOS, hyperprolactinemia, thyroid disease, hypothalamic causes |
CATEGORY 1: Amenorrhea WITHOUT Secondary Sexual Characteristics
A. Hypergonadotropic Hypogonadism (High FSH/LH)
The gonads fail to produce estrogen/inhibin, so negative feedback on the hypothalamic-pituitary axis is lost, causing elevated gonadotropins. ~30% of patients with primary amenorrhea have a karyotypic abnormality.
Turner Syndrome (45,X) - most common cause in this group
- Initially normal ovarian development in utero, then accelerated follicular atresia → streak (fibrotic) ovaries
- Stigmata: short stature, webbed neck, shield chest, cubitus valgus, low hairline, high-arched palate, multiple pigmented nevi, short 4th metacarpal
- Associated conditions: coarctation of the aorta (30%), horseshoe kidney, autoimmune thyroiditis, hearing loss, diabetes
- If Y cell line present (45,X/46,XY mosaicism): gonadectomy required due to risk of gonadoblastoma and dysgerminoma
- Pregnancy via oocyte donation is possible but carries risk of aortic dissection - requires careful cardiac evaluation beforehand
Other chromosomal/genetic causes:
- Partial X chromosome deletions (46,XX with deletions): variable phenotype overlapping Turner syndrome
- Pure gonadal dysgenesis: 46,XX or 46,XY (Swyer syndrome) individuals with streak gonads; those with XY must undergo gonadectomy
- 17α-hydroxylase deficiency: enzyme defect prevents normal estrogen production; patients have streak gonads, absent sex characteristics, hypertension, and hypokalemia
- FSH receptor mutation: autosomal recessive single amino acid substitution (described in Finland); prevents FSH binding; variable secondary sexual development; high FSH/LH
- Gonadal damage before puberty: radiation, alkylating agents (e.g., cyclophosphamide), or combined chemo-radiation
B. Hypogonadotropic Hypogonadism (Low/Normal FSH/LH)
The hypothalamus or pituitary fails to drive the ovary, so estrogen remains low. Gonads are intact but unstimulated.
Physiologic (Constitutional) Delay - most common cause in this group
- GnRH pulse generator is delayed in reactivation
- FSH/LH are functionally deficient for chronologic age but normal for physiologic age
- Family history of delayed puberty common
- Management: reassurance; development will eventually occur
Kallmann Syndrome - second most common hypothalamic cause
- Deficient pulsatile GnRH secretion due to failure of GnRH neuron migration during fetal development
- Classic association: anosmia (though the patient may not be aware of it)
- Varied modes of genetic transmission (X-linked, autosomal dominant, autosomal recessive)
- Treatment: hormone replacement therapy; pulsatile GnRH or injectable gonadotropins for ovulation induction
GnRH receptor mutations
- Autosomal recessive; most patients are compound heterozygotes
- Mutations impair GnRH binding or second-messenger signaling → no FSH/LH stimulation
- Patients are normosomic (distinguishes from Kallmann syndrome)
5α-Reductase Deficiency (46,XY)
- Testosterone cannot be converted to dihydrotestosterone (DHT)
- No breast development (testosterone suppresses it; DHT is required for male external genitalia)
- Normal wolffian duct derivatives (testosterone-dependent) but absent external male genitalia
- Low gonadotropins (testosterone maintains feedback)
FSH Deficiency
- Rare; presents with primary amenorrhea and delayed puberty
- Distinguishing feature: decreased FSH but increased LH (opposite of hypergonadotropic pattern)
- Autosomal recessive mutations in FSHβ subunit
- Treatment: injectable gonadotropins (pregnancy reported)
Other CNS/hypothalamic causes:
- Craniopharyngioma (most common CNS tumor causing primary amenorrhea) - extracellular mass interfering with GnRH/pituitary gonadotropin secretion; virtually all have multi-hormone deficiencies
- Functional causes (malnutrition, anorexia nervosa, excessive exercise, chronic disease, marijuana use) - more commonly cause secondary amenorrhea but can occasionally present as primary amenorrhea if the problem predates puberty
- Hypothyroidism, PCOS, Cushing syndrome, hyperprolactinemia - rare causes of primary amenorrhea
CATEGORY 2: Amenorrhea WITH Secondary Sexual Characteristics + Abnormal Pelvic Anatomy
Congenital abnormalities of the female reproductive organs account for approximately 20% of primary amenorrhea cases. Ovarian function is normal; estrogen is produced; breasts develop. But menstrual outflow is obstructed or absent.
Transverse Outflow Obstruction
- Imperforate hymen
- Transverse vaginal septum
- Absence of cervix or vagina
- These cause cyclic pelvic pain without bleeding; blood accumulates causing hematocolpos, hematometra, or hemoperitoneum, and may lead to endometriosis
- Diagnosis: bulging bluish membrane at introitus (imperforate hymen); MRI for higher obstructions
- Treatment: surgical correction
Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome
- Vaginal agenesis with variable uterine development; karyotype 46,XX
- Accounts for 10-15% of all primary amenorrhea
- Two subtypes:
- Type 1: isolated Müllerian aplasia
- Type 2: Müllerian anomaly + renal malformations (absent/horseshoe kidney, double collecting system), skeletal abnormalities, cardiac defects, hearing impairment
- External genitalia and ovaries are normal; breasts are normal
- Distinction from androgen insensitivity: MRKH has pubic/axillary hair (androgen-sensitive)
- Treatment: vaginal dilation (Frank technique) or surgical creation of neovagina
Androgen Insensitivity Syndrome (AIS) - Complete (CAIS)
- Karyotype 46,XY with testosterone levels in the male range
- Androgen receptor is nonfunctional → tissues cannot respond to testosterone or DHT
- Phenotypically female: normal breast development (testosterone is converted to estrogen peripherally)
- Absent or sparse pubic and axillary hair (key clinical clue vs. MRKH)
- Absent uterus and upper vagina (no Müllerian structures because Müllerian inhibiting substance is produced)
- Blind-ending vaginal pouch
- Testes in abdomen or inguinal canal → risk of malignant transformation; gonadectomy recommended after puberty completes (to allow estrogen-induced breast development from gonadal aromatization)
- Treatment: estrogen replacement after gonadectomy; vaginal dilation if needed
17-20 Lyase Deficiency and 17α-Hydroxylase Deficiency (46,XY)
- Enzyme deficiencies in steroidogenesis; absent uterus; karyotype XY
CATEGORY 3: Amenorrhea WITH Secondary Sexual Characteristics + Normal Pelvic Anatomy
These causes overlap significantly with secondary amenorrhea:
- PCOS - most common cause of anovulatory amenorrhea in reproductive-age women
- Hyperprolactinemia - prolactinomas (rarer in childhood)
- Thyroid disease (hypothyroidism or hyperthyroidism)
- Hypothalamic amenorrhea - functional GnRH suppression (exercise, stress, weight loss)
- Absence of functioning endometrium - Asherman syndrome (rare as a cause of primary amenorrhea; more commonly secondary)
Evaluation
Key Diagnostic Tests
- hCG - rule out pregnancy first
- FSH and estradiol - differentiate hypergonadotropic vs. hypogonadotropic hypogonadism
- LH - assess LH:FSH ratio
- Prolactin (PRL)
- TSH
- Anti-Müllerian hormone (AMH) - may be helpful
- Karyotype - mandatory when gonadal failure is present (to identify Y cell line)
- Pelvic ultrasound or MRI - assess uterus, ovaries, anatomy
- If CNS lesion suspected: MRI brain; if craniopharyngioma suspected: CT/MRI
Evaluation When Secondary Sexual Characteristics Are Absent
- Physical examination - look for Turner stigmata; assess smell (Kallmann); visual fields/optic discs (CNS lesion); galactorrhea
- Short stature with consistent growth + family history → physiologic delay
- Headaches, visual changes, symptoms of diabetes insipidus → CNS lesion
- High FSH → karyotype (Turner, Y line, pure gonadal dysgenesis)
- Low/normal FSH + absent uterus → 5α-reductase deficiency, 17-20 lyase deficiency, 17α-hydroxylase deficiency (all associated with XY karyotype)
- Low/normal FSH + normal uterus → Kallmann syndrome, physiologic delay, disorders of low estrogen before puberty
Treatment Principles
1. Hormone Replacement Therapy (Hypogonadism without Secondary Characteristics)
- Sequential estrogen-progestogen therapy to induce and maintain secondary sexual characteristics
- Typically start with low-dose estrogen, gradually increasing over 2-3 years to mimic normal pubertal progression
- Add cyclic progestogen once breast development is established or after 2 years
- Goals: develop secondary sexual characteristics; protect bone density; provide symptomatic relief of estrogen deficiency
2. Bone Protection
- Calcium and vitamin D supplementation in addition to hormone therapy for women with low circulating estrogen
3. Physiologic Delay
- Reassurance only; development will occur eventually
4. Kallmann Syndrome and Other Hypothalamic Causes
- Hormone replacement therapy
- For fertility: pulsatile GnRH infusion or injectable gonadotropins (clomiphene is ineffective in deeply hypoestrogenic patients)
5. Gonadectomy Indications
- Any patient with a Y cell line in the karyotype (gonadoblastoma risk)
- Turner syndrome 45,X/46,XY mosaicism
- Pure gonadal dysgenesis 46,XY
- Complete AIS (CAIS): typically deferred until after spontaneous puberty, then performed
6. Outflow Obstruction (Imperforate Hymen, Septum)
- Surgical correction (hymenectomy, septum resection)
7. MRKH Syndrome
- Vaginal dilation (progressive dilator therapy; Frank technique) is first-line
- Surgical neovagina creation (McIndoe procedure) if dilation fails
- Pregnancy: not possible without uterus; gestational surrogacy is an option
8. Fertility in Turner Syndrome
- Oocyte donation is generally successful
- However: aortic dissection during pregnancy is a serious risk - thorough cardiac assessment (echo, MRI aorta) mandatory before pursuing pregnancy with donated oocytes
Key Points Summary
| Point | Detail |
|---|---|
| Definition | No menses by age 13 (no secondary sexual characteristics) or age 15 (with normal secondary sexual characteristics) |
| ~30% of cases | Associated karyotypic abnormality when gonadal failure is present |
| Most common hypergonadotropic cause | Turner syndrome (45,X) |
| Most common hypothalamic cause | Physiologic (constitutional) delay |
| Second most common hypothalamic cause | Kallmann syndrome (+ anosmia) |
| 20% of cases | Caused by congenital reproductive organ abnormalities (outflow/Müllerian) |
| 10-15% of cases | Müllerian agenesis (MRKH syndrome) |
| Y cell line → | Gonadectomy to prevent gonadoblastoma |
| CAIS vs. MRKH | Both: absent uterus, female phenotype, 46,XY vs. 46,XX - key difference: pubic/axillary hair absent in CAIS, present in MRKH |
Berek & Novak's Gynecology, Chapter 34 (Amenorrhea), pp. 1856-1873
This is a shared conversation. Sign in to Orris to start your own chat.