Create note on o cardiac attack clinical cardio for 15 marks mgr university bpt exam 2026
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full 15-mark BPT exam answer.
Cardiac Attack (Myocardial Infarction) - Clinical Cardiology
MGR University BPT Exam 2026 | 15-Mark Answer
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of the heart muscle resulting from ischemia. The 2018 joint task force of U.S. and European Cardiology groups defines MI as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- Robbins & Kumar Basic Pathology, p. 353
Incidence and Epidemiology
- Approximately 10% of MIs occur before 40 years of age; 45% occur before 65 years
- Men are at greater risk than women, though the gap narrows with age
- Women are relatively protected during reproductive years, but menopause (with declining estrogen) is associated with increased risk
- IHD (ischemic heart disease) remains the most common cause of death in older adult women
Etiopathogenesis
Primary Cause: Atherosclerotic Plaque Disruption
The vast majority of MIs are caused by acute thrombosis within coronary arteries following disruption of an atherosclerotic plaque. The typical sequence:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and are activated - releasing thromboxane A2, ADP, and serotonin, which cause further platelet aggregation and vasospasm
- Activation of coagulation by tissue factor exposure adds to the growing thrombus
- Within minutes, the enlarging thrombus completely occludes the coronary artery lumen
Angiography within 4 hours of MI onset demonstrates coronary thrombosis in almost 90% of cases.
Other Causes (10% of MIs)
- Coronary artery vasospasm
- Embolism from mural thrombi (e.g., in atrial fibrillation) or valve vegetations
- Prolonged increased demand in setting of severe fixed coronary atherosclerosis (e.g., tachycardia, hypertension)
- Disorders of small intramyocardial arterioles: vasculitis, amyloid deposition, sickle cell disease
Pathophysiology
Immediate Effects of Coronary Occlusion
- Blood flow ceases beyond the occlusion; cardiac muscle requires ~1.3 mL O2/100 g/min just to survive (normal resting delivery: ~8 mL/100 g/min)
- Within seconds: aerobic metabolism ceases → ATP drops → lactic acid accumulates → loss of contractility
- 20-40 minutes: if ischemia persists → irreversible damage and coagulative necrosis
Subendocardial Susceptibility
The subendocardial zone is most vulnerable because:
- It is the last area to receive blood from epicardial vessels
- Exposed to high intramural pressures during systole, impeding blood inflow
- Has higher oxygen consumption
With prolonged ischemia, a "wavefront of cell death" moves outward from the subendocardium toward the epicardium.
Systolic Stretch
When normal portions of the ventricle contract, ischemic/dead segments are pushed outward by elevated intraventricular pressure rather than contracting inward. This reduces overall pumping efficiency and is a key mechanism of decreased cardiac output.
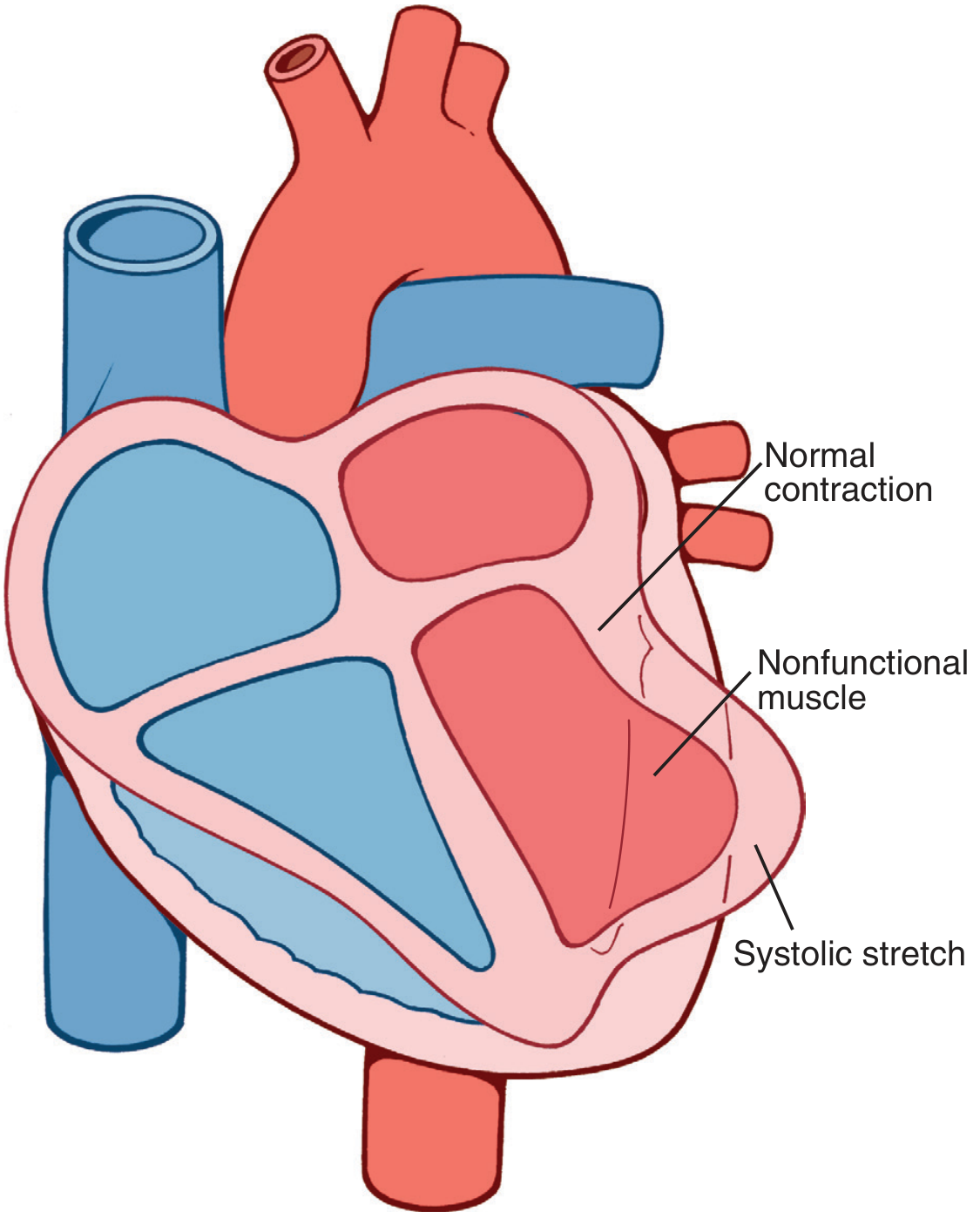
Systolic stretch: nonfunctional ischemic muscle bulges outward while normal myocardium contracts - Guyton & Hall Medical Physiology
Clinical Features
Symptoms
- Chest pain: Severe, crushing, substernal chest pain or pressure - the hallmark symptom
- Radiates to neck, jaw, epigastrium, or left arm
- Lasts several minutes to hours (unlike angina, which lasts <20 min)
- Not relieved by nitroglycerin or rest
- Dyspnea: Due to impaired myocardial contractility and acute pulmonary edema
- Nausea and vomiting: Especially common with posterior wall MIs
- Diaphoresis (profuse sweating)
Note: ~25% of MIs are "silent" - entirely asymptomatic, especially in diabetics (autonomic neuropathy) and the elderly.
Signs
| Sign | Mechanism |
|---|---|
| Rapid, weak pulse | Reduced cardiac output |
| Hypotension | Pump failure |
| Diaphoresis | Sympathetic activation |
| S3/S4 gallop | Ventricular dysfunction |
| Cardiogenic shock | >40% LV involvement |
ECG Changes
Three major ECG abnormalities occur in acute MI:
| Defect in Infarcted Cells | Current Flow | ECG Change (Leads over Infarct) |
|---|---|---|
| Rapid repolarization (accelerated K+ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (K+ loss) | Into infarct | TQ segment depression → recorded as ST segment elevation |
| Delayed depolarization | Out of infarct | ST segment elevation |
- Hallmark of acute MI: ST segment elevation in leads overlying the infarct area
- Leads on opposite side of heart show ST segment depression (reciprocal changes)
- After days/weeks: Q waves develop (pathological) as dead muscle becomes electrically silent
- T wave inversions: Represent abnormalities in myocardial repolarization
STEMI vs. NSTEMI
| Feature | STEMI | NSTEMI |
|---|---|---|
| Occlusion | Complete coronary occlusion | Partial/no complete occlusion |
| Infarct depth | Transmural | Subendocardial/non-transmural |
| ECG | ST elevation + Q waves | ST depression / T wave changes |
| Management | Urgent thrombolysis/PCI | Conservative ± PCI |
Morphological Changes (Timeline)
| Time Frame | Gross Features | Microscopic Features |
|---|---|---|
| 0-30 min | None | None (reversible injury) |
| 30 min - 4 hrs | None | Sarcolemmal disruption; mitochondrial densities |
| 4-12 hrs | Occasional dark mottling | Coagulation necrosis begins; edema; hemorrhage |
| 12-24 hrs | Dark mottling | Ongoing coagulation necrosis; pyknosis of nuclei; hypereosinophilic myocytes; early neutrophilic infiltrate |
| 1-3 days | Mottling with yellow-tan center | Extensive coagulation necrosis; loss of nuclei; macrophage infiltration |
| 3-7 days | Hyperemic border; yellow-tan softening | Phagocytosis of necrotic cells; granulation tissue begins |
| 1-3 weeks | Grey-white scar begins | Fibrosis (scarring) |
| >2 months | White, firm scar | Dense collagenous scar |
- Robbins & Kumar Basic Pathology, Table 9.2
Cardiac Biomarkers (Lab Diagnosis)
Intracellular proteins leak out through damaged sarcolemmal membranes:
| Biomarker | Rises | Peaks | Normalizes | Notes |
|---|---|---|---|---|
| Troponin T / I | 3-4 hrs | 24-48 hrs | 1-2 weeks | Most specific and sensitive; gold standard |
| CK-MB | 4-6 hrs | 18-24 hrs | 48-72 hrs | Useful for reinfarction detection |
| Myoglobin | 1-2 hrs | 4-6 hrs | 24 hrs | Earliest marker; not cardiac specific |
| LDH | 24-48 hrs | 3-5 days | 1-2 weeks | Used when patient presents late |
Troponins have the highest specificity and sensitivity for myocardial damage.
Causes of Death After MI
The four major causes of death (Guyton & Hall, p. 271):
- Decreased cardiac output (cardiogenic shock via systolic stretch)
- Pulmonary edema - damming of blood in pulmonary vessels
- Ventricular fibrillation - most common cause overall (80-90% of MI deaths before hospitalization); due to electrical instability of ischemic myocardium
- Cardiac rupture (rare but catastrophic)
Complications
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Hours to days | VF most common cause of pre-hospital death |
| Cardiogenic shock | Acute | >40% LV damage; high mortality |
| Cardiac rupture | 5-14 days | Softening of necrotic muscle; pericardial tamponade |
| Papillary muscle rupture | 2-7 days | Acute mitral regurgitation |
| Ventricular aneurysm | Weeks to months | Paradoxical wall motion; thromboembolism |
| Dressler syndrome | 2-10 weeks | Autoimmune pericarditis |
| Heart failure | Chronic | Loss of functional myocardium |
| Stunned myocardium | Post-reperfusion | Transient contractile dysfunction despite restored flow |
Management Principles
Immediate (Emergency)
- MONA protocol: Morphine, Oxygen, Nitrates, Aspirin
- Aspirin (300 mg stat) + Clopidogrel (antiplatelet)
- Thrombolysis (tPA, streptokinase) if PCI not available within 90 min
- Primary PCI (Percutaneous Coronary Intervention) - gold standard for STEMI; early angioplasty + stent placement limits infarct size
Medical
- Beta-blockers: Reduce heart rate, myocardial O2 demand
- ACE Inhibitors / ARBs: Prevent ventricular remodeling
- Statins: Plaque stabilization and lipid lowering
- Anticoagulants (heparin, LMWH): Prevent further thrombosis
Rationale for Early Intervention
Since early thrombolysis/angioplasty can be highly successful in limiting the extent of myocardial necrosis (ischemic injury is reversible if reperfused before 20-40 min), time-to-treatment is the most critical factor in outcome.
Physiotherapy Relevance (BPT Focus)
- Physiotherapists play a key role in Cardiac Rehabilitation post-MI
- Phase I (in-hospital): Early ambulation, breathing exercises, positioning
- Phase II/III (outpatient): Graded aerobic exercise (target 50-70% max HR), progressive walking, cycle ergometry
- Monitor for anginal symptoms, arrhythmias, hypotension during exercise
- Contraindications to exercise: Unstable angina, uncontrolled arrhythmias, cardiogenic shock, acute MI within 48 hrs
- Education: Risk factor modification - smoking cessation, weight management, dietary control
Summary
| Aspect | Key Points |
|---|---|
| Definition | Myocardial necrosis due to ischemia |
| Main cause | Atherosclerotic plaque rupture → coronary thrombosis |
| Hallmark symptom | Crushing substernal chest pain, not relieved by rest |
| Key ECG change | ST elevation (STEMI) or ST depression (NSTEMI) |
| Best biomarker | Troponin T/I |
| Most common cause of death | Ventricular fibrillation |
| Gold standard treatment | Primary PCI within 90 minutes |
Sources:
- Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 9 - Ischemic Heart Disease, pp. 353-359
- Guyton and Hall Textbook of Medical Physiology, Chapter 21 - Myocardial Infarction, pp. 271-274
- Ganong's Review of Medical Physiology (26th ed.), Chapter 29 - ECG in Acute MI, pp. 534-536
- Braunwald's Heart Disease - A Textbook of Cardiovascular Medicine
This is a shared conversation. Sign in to Orris to start your own chat.