Anemia
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Finding Sources
Reading File
Loading Image
I now have all the key content I need from Robbins. Let me compile the comprehensive exam-focused study guide.
Sickle Cell Disease & Thalassemia - Exam Study Guide
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
SICKLE CELL DISEASE
Genetics & Molecular Basis
- Mutation: Single missense point mutation in the β-globin gene (chromosome 11) - glutamate replaced by valine at position 6
- This converts HbA (α₂β₂) to HbS (α₂β^S₂)
- Inheritance: Autosomal recessive
- Sickle cell trait (HbAS): ~8-10% of African Americans; ~40% HbS, rest HbA - largely asymptomatic; HbA interferes with HbS polymerization
- Sickle cell disease: Homozygous HbSS; ~70,000-100,000 affected in the US
- Malaria protection: HbS mutation arose in West Africa; provides protection against P. falciparum malaria by promoting clearance of parasitized cells and impairing PfEMP-1 membrane knob formation
Pathophysiology
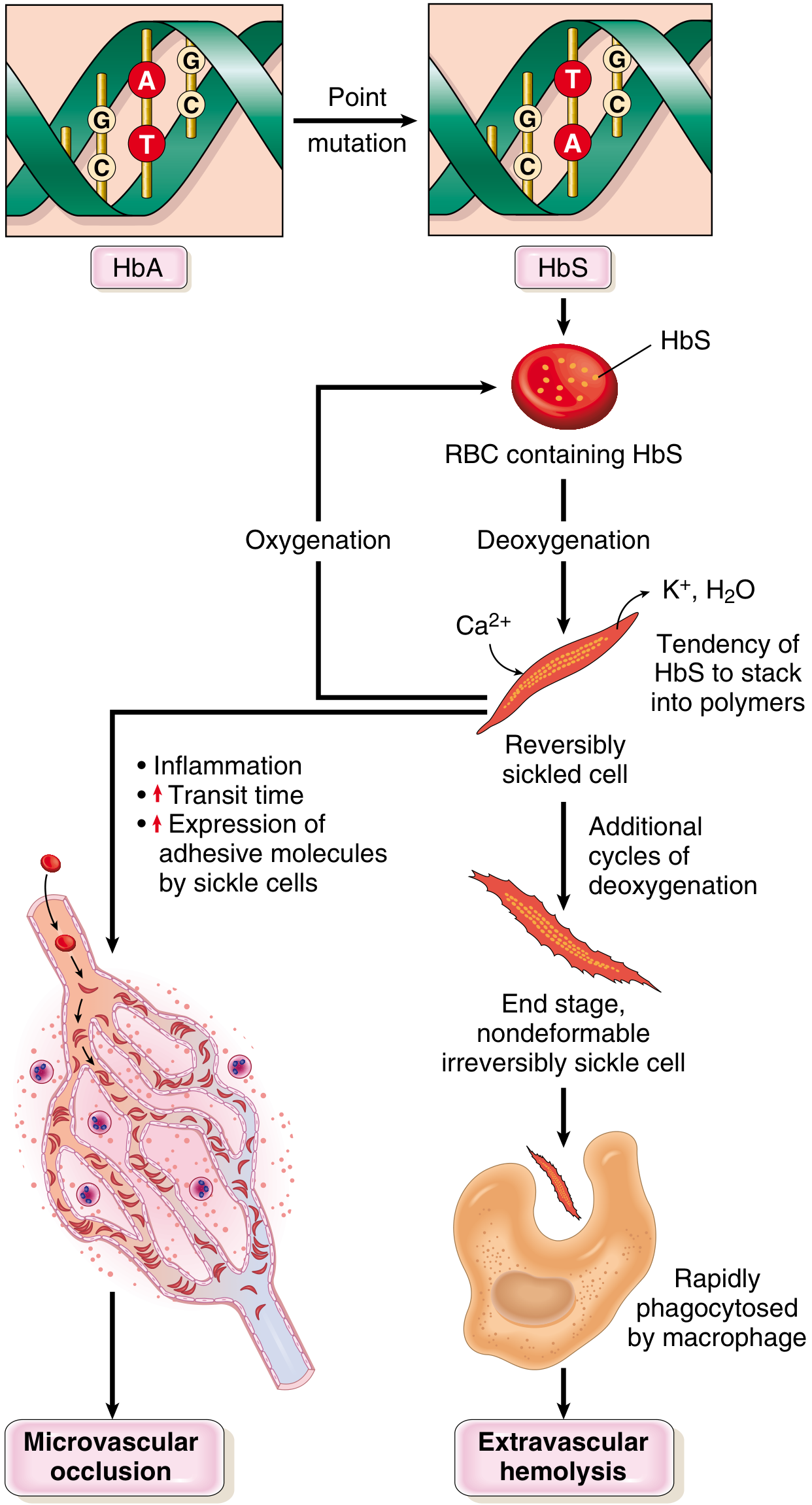
Key mechanism: Deoxygenated HbS molecules polymerize into long needlelike fibers → red cell distortion into sickle/holly-leaf shape
Factors that increase sickling:
| Factor | Effect |
|---|---|
| Low O₂ tension | Core trigger - deoxygenated HbS polymerizes |
| High HbS concentration / dehydration (high MCHC) | More aggregation |
| Low pH / acidosis | Reduces O₂ affinity → more deoxyHbS |
| Slow microvascular transit | Prolonged deoxygenation (spleen, bone marrow) |
| Inflammation | Up-regulates endothelial adhesion molecules |
| HbC co-inheritance | More sickling than HbAS |
Factors that decrease sickling:
- HbF (fetal hemoglobin) - interferes with polymerization
- Co-existing α-thalassemia (reduces MCHC)
- HbA (in heterozygotes)
Downstream cascade:
- Repeated sickling → Ca²⁺ influx → K⁺ & H₂O efflux → dehydrated, dense, rigid cells
- Irreversibly sickled cells (ISCs) form - retain sickle shape even when oxygenated
- ISCs cleared by extravascular hemolysis (spleen/liver macrophages)
- Intravascular hemolysis releases free Hb → binds/inactivates NO → vasoconstriction + platelet aggregation → vascular occlusion
- Sickle cells express increased adhesion molecules → adhere to endothelium → microvascular occlusion
Morphology
- Peripheral blood smear: Irreversibly sickled cells, target cells, reticulocytosis, Howell-Jolly bodies (nuclear remnants, due to functional asplenia)
- Bone marrow: Hyperplastic (compensatory erythroid hyperplasia)
- Spleen in children: Enlarged (up to 500g), red pulp congested with trapped sickled cells
- Spleen in adults: Small fibrotic nubbin = autosplenectomy (repeated infarctions)
Clinical Features
Baseline anemia: Hematocrit 18-30%; moderate hemolytic anemia, reticulocytosis, hyperbilirubinemia
Crises (high-yield exam topics):
| Crisis Type | Mechanism | Key Features |
|---|---|---|
| Vaso-occlusive (pain) crisis | Ischemic infarction from sickling in microvasculature | Most common; fever, severe pain; bones, lungs, liver, brain, spleen, penis |
| Acute chest syndrome | Vaso-occlusion in pulmonary vessels | Fever, cough, chest pain, pulmonary infiltrates; potentially fatal; requires exchange transfusion |
| Sequestration crisis | Massive splenic trapping of sickled RBCs | Rapid splenomegaly, hypovolemia, shock; children with intact spleens |
| Aplastic crisis | Parvovirus B19 infects erythroid progenitors | Sudden severe anemia; transient cessation of erythropoiesis |
Organ complications:
- Dactylitis / hand-foot syndrome: Painful bone crises in small bones of hands/feet in infants/children - often first manifestation
- Stroke: Due to adhesion of sickled RBCs to arterial endothelium + NO depletion causing vasoconstriction
- Priapism: Up to 45% of males after puberty; can cause erectile dysfunction
- Retinopathy: Vascular occlusion; may cause blindness
- Leg ulcers: Adults; subcutaneous vascular stagnation
- Hyposthenuria: Renal medullary damage (sickling promoted by hypertonicity) → inability to concentrate urine → dehydration risk
- Infections: Encapsulated organisms (especially Streptococcus pneumoniae, H. influenzae) due to functional asplenia + complement pathway defects
Diagnosis
- Peripheral smear: sickled cells
- Hemoglobin electrophoresis: Confirms HbS; differentiates trait vs. disease
- Sickling tests (metabisulfite test)
- Newborn screening: routine in all 50 US states (heel stick)
- Prenatal diagnosis: fetal DNA from amniocentesis or chorionic biopsy
Treatment
- Hydroxyurea (mainstay): Inhibits DNA synthesis; benefits include:
- Increases HbF synthesis (HbF inhibits sickling)
- Decreases adhesion molecule expression on RBCs
- Reduces WBC count (anti-inflammatory)
- Reduces frequency of painful crises and acute chest syndrome; improves survival
- Exchange transfusion: For acute chest syndrome, stroke
- Bone marrow / stem cell transplant: Only curative option
- Supportive: Hydration, analgesics, vaccinations, prophylactic penicillin in children
- Gene therapy: Emerging curative approach
THALASSEMIA
Overview
Inherited disorders with decreased synthesis of α- or β-globin chains. Double problem:
- Insufficient HbA → microcytic, hypochromic anemia
- Excess unpaired globin chains → precipitates → RBC membrane damage → hemolysis
Particularly common in Mediterranean, African, and Asian populations (malaria-endemic regions).
β-Thalassemia
Genetics
- Gene: β-globin gene, chromosome 11 (single gene per haploid)
- Mutations: >100 mutations, mostly point mutations (not deletions) → affect transcription, mRNA splicing, or translation
- β⁰: No β-globin produced
- β⁺: Reduced β-globin produced
Classification
| Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia major (Cooley's anemia) | β⁰/β⁰ or β⁰/β⁺ (homozygous) | Severe anemia; requires regular transfusions |
| β-Thalassemia intermedia | Variable β⁺/β⁺ or mild β⁰/β⁺ | Moderate anemia; transfusions NOT regularly required |
| β-Thalassemia minor (trait) | Heterozygous β/β⁰ or β/β⁺ | Asymptomatic or mild; red cell abnormalities on smear |
Pathophysiology
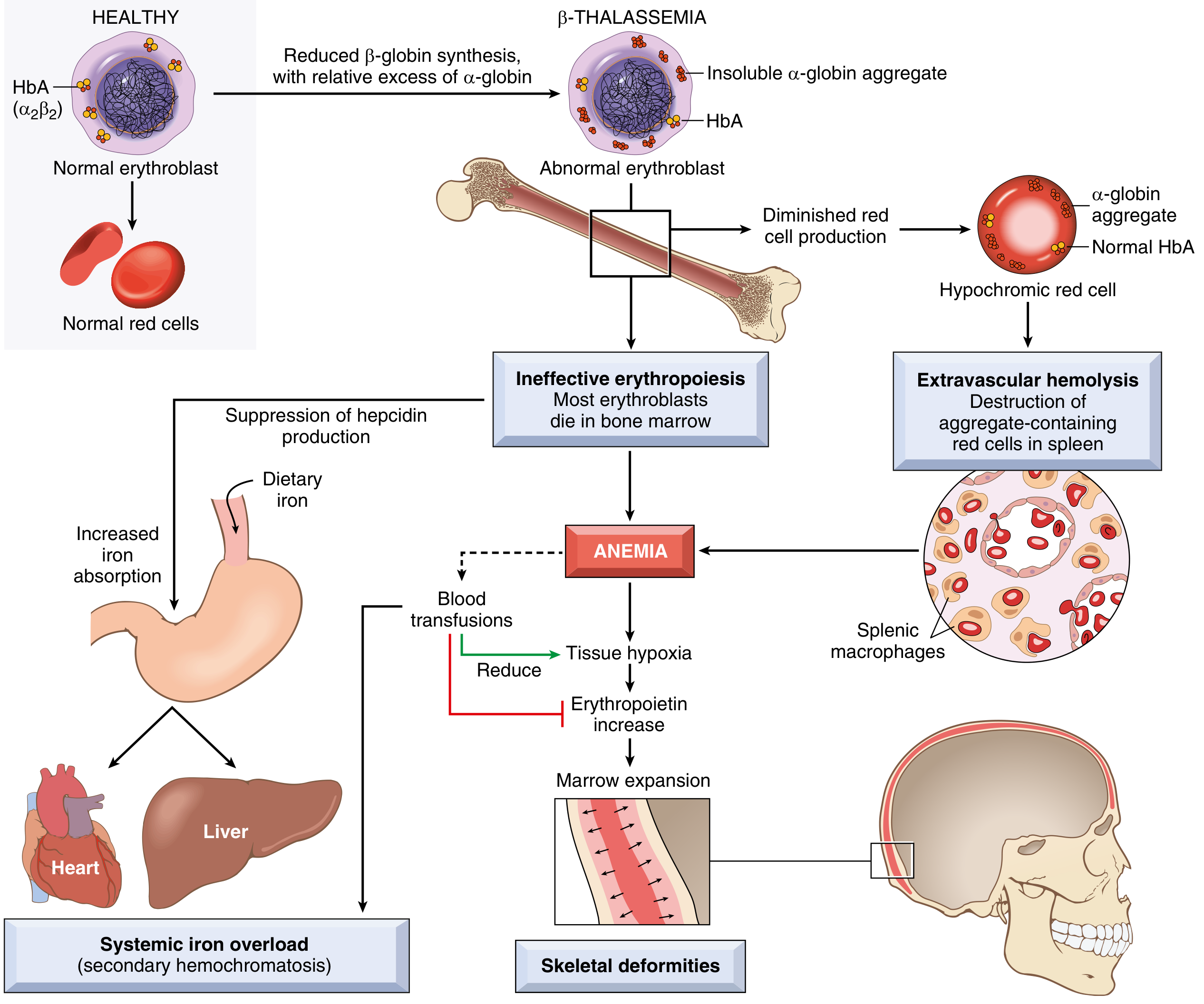
- Reduced β-globin → relative excess of α-globin chains
- Excess α-chains form toxic insoluble precipitates that damage erythroid precursor membranes → apoptosis in bone marrow = ineffective erythropoiesis (most important mechanism)
- Few surviving RBCs have shortened lifespan → extravascular hemolysis in spleen
- Anemia → ↑ erythropoietin → massive erythroid marrow expansion → bone deformities
- Low hepcidin levels → increased dietary iron absorption → secondary hemochromatosis (even without transfusions)
- Transfusions worsen iron overload
Clinical Features of β-Thalassemia Major
- Presents 6-12 months after birth (when HbF→HbA switch occurs)
- Severe anemia: pallor, jaundice, hepatosplenomegaly
- Skeletal deformities: Erythroid marrow expansion causes:
- "Crew-cut" skull X-ray appearance (hair-on-end pattern)
- Frontal bossing, maxillary hypertrophy ("chipmunk facies")
- Thinning of cortical bone → pathological fractures
- Growth retardation, failure to thrive
- Iron overload (secondary hemochromatosis):
- Cardiac failure (leading cause of death)
- Cirrhosis
- Endocrine failure (diabetes, hypogonadism)
- Increased susceptibility to infections
Treatment of β-Thalassemia Major
- Regular blood transfusions (maintain Hb >9-10 g/dL)
- Iron chelation therapy: Deferoxamine (IV/SC) or deferasirox (oral) - mandatory to prevent iron overload
- Splenectomy: If hypersplenism causing excessive transfusion needs
- Bone marrow / stem cell transplant: Curative; best results in young patients
- Hydroxyurea: Can increase HbF in some patients
α-Thalassemia
Genetics
- Gene: α-globin genes - TWO genes per chromosome 16 (total 4 α-globin genes)
- Mutations: Mostly gene deletions (contrast with β-thalassemia which is mostly point mutations)
Classification (by number of deleted α genes)
| # Genes Deleted | Syndrome | Genotype | Clinical Features |
|---|---|---|---|
| 1 | Silent carrier | -α/αα | Asymptomatic; normal CBC |
| 2 | α-Thalassemia trait | --/αα (Asian) or -α/-α (African/Asian) | Asymptomatic; mild microcytic anemia (like β-thal minor) |
| 3 | HbH disease | --/-α | Moderately severe hemolytic anemia; HbH = β₄ tetramers (unstable); resembles β-thal intermedia |
| 4 | Hydrops fetalis | --/-- | Lethal in utero; Hb Barts (γ₄ tetramers); fetus cannot deliver O₂; stillbirth or death shortly after birth |
Key exam point: With loss of α-globin genes, excess β-globin forms HbH (β₄), and excess γ-globin in fetal life forms Hb Barts (γ₄) - both have very high O₂ affinity and cannot deliver O₂ to tissues.
HIGH-YIELD COMPARISON TABLE
| Feature | Sickle Cell Disease | β-Thalassemia Major | α-Thalassemia Trait |
|---|---|---|---|
| Gene | β-globin (chr 11) | β-globin (chr 11) | α-globin (chr 16) |
| Mutation type | Point mutation (missense) | Point mutations | Gene deletions |
| RBC morphology | Sickle cells, target cells, Howell-Jolly | Target cells, nucleated RBCs, anisopoikilocytosis | Mild microcytosis |
| MCV/MCH | Usually normal/high (with reticulocytosis) | Low (microcytic, hypochromic) | Low (microcytic) |
| Reticulocytes | High | High (but ineffective) | Normal/slightly high |
| Spleen | Autosplenectomy in adults | Massive splenomegaly | Normal |
| Iron stores | Normal to low | Overloaded (even without transfusions) | Normal |
| HbF effect | Protective - decreases sickling | Partially compensatory | N/A |
| Diagnosis | Hb electrophoresis | Hb electrophoresis + elevated HbA2 | Hb electrophoresis + gene testing |
| Definitive cure | Stem cell transplant / gene therapy | Stem cell transplant | Usually none needed |
QUICK MNEMONICS
Sickle cell crises - "PASA":
- Pain (vaso-occlusive) - most common
- Acute chest syndrome - most dangerous
- Sequestration - spleen trapping in children
- Aplastic - parvovirus B19
Hydroxyurea mechanism - "4F's":
- Increases Fetal Hb (HbF)
- Reduces adhesion molecule Forces
- Fewer WBCs (anti-inFlammatory)
- Fewer painful crises (fewer Flare-ups)
α-thalassemia severity: 1-2-3-4 deleted genes = Silent-Trait-HbH-Hydrops (lethal)
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 599-601; Robbins & Kumar Basic Pathology, pp. 387-389
This is a shared conversation. Sign in to Orris to start your own chat.