Thalasimia
Reading File
search_medical_text
thalassemia types pathophysiology clinical features
search_medical_text
thalassemia diagnosis treatment management
search_medical_image
thalassemia blood smear peripheral film
GradeResponse
GradeResponse
GradeResponse
Thalassemia
Overview
Thalassemia is an inherited hemoglobinopathy caused by reduced or absent synthesis of one or more globin chains (α or β), leading to unbalanced globin production. The unpaired excess chains precipitate within developing erythroblasts, damaging them and causing ineffective erythropoiesis and hemolytic anemia (Harrison's, p. 2925).
It is one of the most common single-gene disorders worldwide — an estimated 1–5% of the global population carries a thalassemia mutation, with highest prevalence in Mediterranean, Middle Eastern, South/Southeast Asian, and sub-Saharan African populations. Heterozygotes are partially protected from Plasmodium falciparum malaria, which explains its high frequency in tropical regions.
Types
🔴 Alpha (α) Thalassemia
Caused by deletion or mutation of α-globin genes (normally 4 copies: αα/αα).
| Genotype | Name | Features |
|---|---|---|
| 3 genes intact (-α/αα) | Silent carrier | Asymptomatic, normal CBC |
| 2 genes intact (--/αα or -α/-α) | α-Thal trait | Mild microcytic anemia |
| 1 gene intact (--/-α) | HbH disease | Moderate–severe hemolytic anemia, splenomegaly |
| 0 genes intact (--/--) | Hydrops fetalis (Hb Bart's) | Incompatible with life; fetal death |
🔵 Beta (β) Thalassemia
Caused by point mutations in the β-globin gene (chromosome 11), reducing (β⁺) or abolishing (β⁰) β-chain synthesis.
| Type | Genotype | Features |
|---|---|---|
| β-Thal minor (trait) | β/β⁺ or β/β⁰ | Mild microcytic anemia, usually asymptomatic |
| β-Thal intermedia | β⁺/β⁺ or β⁺/β⁰ | Moderate anemia; may not require regular transfusions |
| β-Thal major (Cooley's anemia) | β⁰/β⁰ | Severe transfusion-dependent anemia from infancy |
Pathophysiology
- Reduced globin synthesis → imbalanced α:β ratio
- Excess unpaired chains precipitate → membrane damage → premature destruction of erythroblasts in bone marrow (ineffective erythropoiesis)
- Surviving RBCs are fragile → hemolytic anemia
- Chronic anemia → erythropoietin surge → massive bone marrow expansion → skeletal deformities, extramedullary hematopoiesis
- Chronic hemolysis + repeated transfusions → iron overload → end-organ damage (liver, heart, endocrine glands)
Clinical Features
β-Thalassemia Major (most severe form)
- Presents at 6–24 months of age (when fetal hemoglobin switches to adult Hb)
- Severe pallor, jaundice, hepatosplenomegaly
- "Thalassemic facies": frontal bossing, prominent cheekbones, maxillary overgrowth (due to marrow expansion)
- Growth retardation, recurrent infections
- Iron overload (from transfusions + increased GI absorption): cardiomyopathy, liver cirrhosis, diabetes, hypogonadism, hypothyroidism
β-Thalassemia Intermedia
- Symptomatic but not transfusion-dependent from birth
- Splenomegaly, extramedullary hematopoietic masses
- Leg ulcers, thrombotic complications
β-Thalassemia Minor (Trait)
- Usually asymptomatic; detected incidentally
- Mild microcytic hypochromic anemia (must be differentiated from iron deficiency)
Peripheral Blood Smear Findings
The image below illustrates classic thalassemia findings:
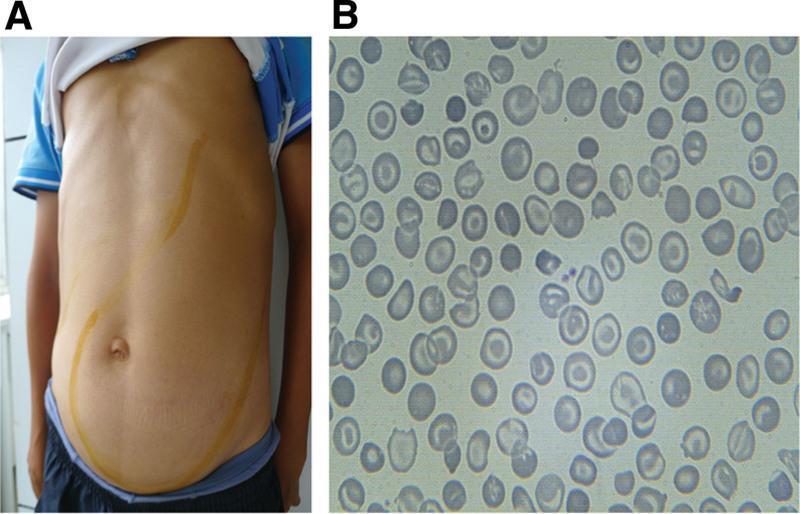
Key smear findings:
- Microcytosis (small RBCs)
- Hypochromia (increased central pallor)
- Target cells (codocytes)
- Schistocytes, poikilocytosis, nucleated RBCs (in severe disease)
Diagnosis
Lab Investigations
| Test | Finding in β-Thal Minor | Finding in β-Thal Major |
|---|---|---|
| CBC | Low MCV (<70 fL), mild anemia | Severe anemia (Hb 3–7 g/dL) |
| Peripheral smear | Microcytes, target cells | Severe dysmorphia, NRBCs |
| Hb electrophoresis / HPLC | ↑ HbA₂ (>3.5%) ± ↑ HbF | Predominantly HbF, absent/reduced HbA |
| Serum ferritin | Normal | Elevated (iron overload) |
| Bilirubin | Normal/mildly ↑ | Elevated (hemolysis) |
| Reticulocyte count | Normal or mildly ↑ | Elevated |
Key diagnostic test: Hemoglobin HPLC or electrophoresis — elevated HbA₂ (>3.5%) is the hallmark of β-thalassemia trait (Harrison's, p. 2928).
Genetic testing is required for carrier identification and prenatal diagnosis (to identify the specific mutation before counseling).
Differentiating from Iron Deficiency Anemia
| Feature | Iron Deficiency | β-Thal Trait |
|---|---|---|
| Serum ferritin | ↓ | Normal |
| HbA₂ | Normal | ↑ (>3.5%) |
| RBC count | ↓ | Normal or ↑ |
| Mentzer index (MCV/RBC) | >13 | <13 |
| Response to iron | Yes | No |
Management
β-Thalassemia Major
1. Regular Blood Transfusions
- Target pre-transfusion Hb: 9–10 g/dL
- Keeps Hb stable, suppresses ineffective erythropoiesis, prevents skeletal deformities
2. Iron Chelation Therapy (mandatory with chronic transfusions)
- Deferoxamine (IV/SC) — standard agent
- Deferasirox (oral) — commonly used
- Deferiprone (oral) — especially effective for cardiac iron
3. Splenectomy
- Considered when transfusion requirements increase significantly
- Risk of post-splenectomy infections (vaccinate against encapsulated organisms)
4. Hematopoietic Stem Cell Transplantation (HSCT)
- Only curative option
- Best outcomes in young patients with HLA-matched sibling donors
- 80–90% disease-free survival in low-risk patients
5. Novel/Emerging Therapies
- Luspatercept (activin receptor ligand trap): reduces transfusion burden in β-thal
- Gene therapy (betibeglogene autotemcel / Zynteglo): approved; introduces functional β-globin gene — potentially curative
- Gene editing (CRISPR/Cas9): fetal hemoglobin (HbF) reactivation strategies under investigation
β-Thalassemia Minor
- No treatment needed
- Genetic counseling essential — if both partners are carriers, each pregnancy has a 25% chance of producing a child with thalassemia major
Folic Acid
- Supplementation recommended in all patients with hemolytic anemia (increased folate consumption)
Complications
| System | Complication |
|---|---|
| Cardiac | Dilated cardiomyopathy (iron), arrhythmias |
| Liver | Cirrhosis, hepatocellular carcinoma |
| Endocrine | Diabetes, hypogonadism, hypothyroidism, growth retardation |
| Bone | Osteoporosis, pathological fractures, thalassemic facies |
| Infectious | Post-splenectomy sepsis, transfusion-transmitted infections |
| Thrombotic | Hypercoagulable state (especially in intermedia) |
Prognosis
- β-Thal minor: Normal life expectancy
- β-Thal major (well-managed): Survival into the 4th–5th decade with good chelation; cardiac iron overload remains the leading cause of death
- HSCT/gene therapy: Offers potential cure with excellent long-term outcomes
Prevention
- Carrier screening (CBC + HPLC in at-risk populations)
- Prenatal diagnosis: chorionic villus sampling (CVS) at 10–12 weeks or amniocentesis
- Preimplantation genetic diagnosis (PGD) in IVF cycles
National programs in countries like Cyprus, Sardinia, and Iran have dramatically reduced thalassemia major births through mass carrier screening.
This is a shared conversation. Sign in to Orris to start your own chat.