Hi
Hey! How can I help you today?
Tell about you
I'm Orris, a knowledgeable AI assistant built to help you across a wide range of topics. Here's a quick overview:
What I Can Do
Medicine & Health
- Answer clinical, pharmacology, anatomy, and physiology questions
- Draw from a library of authoritative medical textbooks
- Search PubMed for peer-reviewed research and current evidence
Research & Information
- Search the web for up-to-date information
- Extract and summarize content from articles and web pages
- Provide well-cited, accurate answers
Documents & Files
- Create and edit Word documents (.docx), PowerPoint presentations (.pptx), Excel spreadsheets (.xlsx), and PDFs
- Read and analyze files you upload
Code & Computation
- Write and execute code (Python, shell scripts, etc.)
- Perform data analysis and calculations
Specialized Knowledge
- Indian taxation (direct taxes, income tax, GST, CA/CMA exam prep)
- ACCA exam preparation
- Orthopaedic surgery (hip procedures and guidelines)
How I Work
- I give you a quick answer first, then refine it with deeper research
- I use parallel tool calls to get you results faster
- I cite sources so you can verify what I tell you
- I'm honest - if I don't know something, I'll say so rather than guess
What would you like to explore? I'm ready to help!
What are the books you use for medicine
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Here is the full list of medical and clinical textbooks in my library, organized by specialty:
Anatomy
- Color Atlas of Ultrasound Anatomy, 3e
- Color Atlas of Human Anatomy Vol. 1, 2 & 3
- General Anatomy and Musculoskeletal System (THIEME Atlas)
- Gray's Anatomy for Students
- Imaging Anatomy - Bones, Joints, Vessels and Nerves
- Netter's Atlas of Human Anatomy
Anesthesiology
- Barash, Cullen & Stoelting's Clinical Anesthesia, 9e
- Miller's Anesthesia, 2-Volume Set, 10e
- Morgan & Mikhail's Clinical Anesthesiology, 7e
Biochemistry
- Basic Medical Biochemistry - A Clinical Approach, 6e
- Biochemistry, 8th ed - Lippincott Illustrated Reviews
- Harper's Illustrated Biochemistry, 32nd Ed
Cardiology
- Braunwald's Heart Disease, 2-Vol Set
- Fuster and Hurst's The Heart, 15th Edition
- Textbook of Clinical Echocardiography
Community Medicine
- Park's Textbook of Preventive and Social Medicine
Dermatology
- Andrews' Diseases of the Skin Clinical Atlas, 2e
- Andrews' Diseases of the Skin - Clinical Dermatology
- Dermatology 2-Volume Set, 5e
- Fitzpatrick's Dermatology, Vol. 1 & 2
Embryology
- Langman's Medical Embryology
- The Developing Human - Clinically Oriented Embryology
Emergency Medicine
- Rosen's Emergency Medicine, 9e
- Roberts and Hedges' Clinical Procedures in Emergency
- Tintinalli's Emergency Medicine - A Comprehensive Study
ENT (Otolaryngology)
- Cummings Otolaryngology Head and Neck Surgery
- K.J. Lee's Essential Otolaryngology
- Scott-Brown's Otorhinolaryngology Head & Neck Surgery (3 vols)
- Shambaugh Surgery of the Ear
Family Medicine
- Pfenninger and Fowler's Procedures for Primary Care, 3e
- Swanson's Family Medicine Review
- Textbook of Family Medicine, 9e
Forensic Medicine
- Brogdon's Forensic Radiology
- DiMaio's Forensic Pathology, 3rd Edition
- Forensic Anthropology - A Comprehensive Introduction, 2e
- P.C. Dikshit Textbook of Forensic Medicine and Toxicology
- Parikh's Textbook of Medical Jurisprudence
- The Essentials of Forensic Medicine and Toxicology, 36th ed (2026)
Gastroenterology
- Clinical Gastrointestinal Endoscopy, 3e
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease
- Yamada's Textbook of Gastroenterology, 7e
General Surgery
- Bailey and Love's Short Practice of Surgery, 28th Edition
- Current Surgical Therapy, 14e
- Fischer's Mastery of Surgery, 8e
- Maingot's Abdominal Operations
- Mulholland and Greenfield's Surgery, 7e
- Pye's Surgical Handicraft, 22nd Edition
- S Das A Manual on Clinical Surgery, 13th Edition
- Sabiston Textbook of Surgery
- Schwartz's Principles of Surgery, 11th Edition
Genetics
- Emery's Elements of Medical Genetics and Genomics
- Thompson & Thompson Genetics and Genomics in Medicine, 9e
Histology
- Histology: A Text and Atlas (Correlated Cell & Molecular Biology)
- Junqueira's Basic Histology, 17e
Immunology
- Cellular and Molecular Immunology
- Janeway's Immunobiology, 10e
- Roitt's Essential Immunology
Internal Medicine
- Firestein & Kelley's Textbook of Rheumatology, 2-Volume Set
- Frameworks for Internal Medicine
- Goldman-Cecil Medicine, 2-Volume Set
- Harrison's Principles of Internal Medicine, 22nd Edition (2025)
- Rheumatology, 2-Volume Set (2022)
- Symptom to Diagnosis - An Evidence Based Guide, 4e
- The Washington Manual of Medical Therapeutics
Laboratory Medicine
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Quick Compendium of Clinical Pathology, 5e
- Tietz Textbook of Laboratory Medicine, 7th Edition
Microbiology
- Jawetz, Melnick & Adelberg's Medical Microbiology, 28e
- Medical Microbiology, 9e
- Sherris & Ryan's Medical Microbiology, 8th Edition
Nephrology
- Brenner and Rector's The Kidney, 2-Volume Set
- Comprehensive Clinical Nephrology, 7th Edition
- NKF Primer on Kidney Diseases, 8e
Neurology
- Adams and Victor's Principles of Neurology, 12th Edition
- Bradley and Daroff's Neurology in Clinical Practice
- Localization in Clinical Neurology, 8e
- Plum and Posner's Diagnosis and Treatment of Stupor and Coma
Neuroscience
- Eric Kandel's Principles of Neural Science, 6th Edition
- Neuroanatomy through Clinical Cases, 3rd Edition
- Neuroscience: Exploring the Brain, 5th Edition
Obstetrics & Gynecology
- Berek & Novak's Gynecology
- Creasy & Resnik's Maternal-Fetal Medicine
Ophthalmology
- Kanski's Clinical Ophthalmology, 10th Edition
- The Wills Eye Manual
Orthopedics
- Campbell's Operative Orthopaedics, 15th Ed (2026)
- Miller's Review of Orthopaedics, 9th Edition
- Rockwood and Green's Fractures in Adults, 10th Ed (2025)
Pathology
- Robbins & Kumar Basic Pathology
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Pediatrics
- Red Book 2021 - Report of the Committee on Infectious Diseases
- The Harriet Lane Handbook, 23rd Edition
Pharmacology
- Goodman & Gilman's The Pharmacological Basis of Therapeutics
- Katzung's Basic and Clinical Pharmacology, 16th Edition
- Lippincott Illustrated Reviews: Pharmacology
Physiology
- Costanzo Physiology, 7th Edition
- Ganong's Review of Medical Physiology, 26th Edition
- Guyton and Hall Textbook of Medical Physiology
- Medical Physiology (Boron & Boulpaep)
Psychiatry
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry
- Kaplan and Sadock's Synopsis of Psychiatry
- Stahl's Essential Psychopharmacology
- The Clozapine Handbook
- The Maudsley Deprescribing Guidelines
- The Maudsley Prescribing Guidelines in Psychiatry, 15th Ed
Pulmonology
- Fishman's Pulmonary Diseases and Disorders, 2-Volume Set
- Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set
Radiology
- Grainger & Allison's Diagnostic Radiology
Urology
- Campbell Walsh Wein Urology, 3-Volume Set
- Hinman's Atlas of Urologic Surgery
- Smith and Tanagho's General Urology, 19th Edition
That's 130+ textbooks spanning virtually every medical specialty. Many are the latest editions (some as recent as 2025-2026). Is there a specific topic or specialty you'd like to ask about?
Treatment for Pre eclampsia
Reading File
Finding Sources
Searching PubMed
"preeclampsia"[MeSH Terms] AND "treatment"[MeSH Terms]
Finding Sources
Searching PubMed
preeclampsia management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information to provide a thorough answer. Let me compile it.
Treatment of Preeclampsia
Preeclampsia is a multisystem disorder of pregnancy defined by new-onset hypertension (BP ≥140/90 mmHg) after 20 weeks' gestation, with or without proteinuria, plus associated features (thrombocytopenia, renal insufficiency, impaired liver function, pulmonary edema, or new-onset headache/visual disturbances). The only definitive cure is delivery.
1. Timing of Delivery
The cornerstone of management is deciding when to deliver:
| Gestational Age | Approach |
|---|---|
| < 24 weeks | Delivery usually recommended - perinatal mortality >80% even with expectant management; maternal complications are common |
| 24-37 weeks | Balance fetal benefit of prolonging pregnancy vs. maternal risk. Expectant management with intensive monitoring may be considered |
| > 37 weeks | Immediate delivery almost always indicated |
Indications for expedient delivery regardless of gestational age:
- Non-reassuring fetal testing
- Suspected placental abruption
- Thrombocytopenia
- Worsening liver or kidney function
- Unrelenting headache, visual changes, nausea/vomiting, or epigastric pain
RCTs have shown that in women with severe preeclampsia between 28-32 weeks, expectant management (delaying delivery 1-2 weeks) decreases neonatal complications and NICU stay without significantly increasing maternal complications. Mode of delivery: induction of labor vs. cesarean section shows similar maternal/neonatal outcomes in retrospective studies.
(Brenner and Rector's The Kidney, p. 2156)
2. Blood Pressure Management
The goal in preeclampsia is not to minimize long-term cardiovascular risk (as in non-pregnant patients), but to maximize safe delivery of a healthy infant while preventing acute maternal complications.
- Threshold to treat: BP > 150-160 mmHg systolic or > 100-110 mmHg diastolic (above this, risk of cerebral hemorrhage becomes significant)
- Aggressive acute lowering of BP is avoided - it can cause fetal distress or demise by compromising placental perfusion
- The CHIPS trial (2015) showed that targeting diastolic BP of 85 mmHg (tight control) vs. 100 mmHg (less-tight) is safe for the fetus and lowers short-term maternal complications (less severe hypertension, thrombocytopenia, transaminitis)
Antihypertensive Agents
First-Line (Oral):
| Drug | Notes |
|---|---|
| Methyldopa | First line; most extensive safety data; centrally acting alpha-2 agonist; short half-life, requires multiple daily dosing |
| Labetalol | Alpha + beta blocker; preferred over other beta-blockers due to beneficial alpha-blockade on uteroplacental flow; avoid in asthma |
| Long-acting Nifedipine | Once-daily dosing; calcium channel blocker; may cause edema |
First-Line (Intravenous) for Acute Severe Hypertension:
| Drug | Notes |
|---|---|
| IV Labetalol | Good safety data |
| IV Nicardipine | Extensive safety data; effective |
Second-Line:
- Hydralazine: extensive clinical experience but increased risk of maternal hypotension and placental abruption with acute use
- Metoprolol, Verapamil, Diltiazem: limited data but no known adverse fetal effects
Generally Avoided:
- Diuretics: may impair pregnancy-associated plasma volume expansion
- Atenolol: associated with fetal growth restriction
- Nitroprusside: risk of fetal cyanide poisoning if used > 4 hours
Contraindicated:
- ACE inhibitors and ARBs: multiple fetal anomalies (renal dysgenesis, oligohydramnios, skull ossification defects)
(Brenner and Rector's The Kidney, Table 48.6, p. 2160)
3. Seizure Prophylaxis - Magnesium Sulfate
Magnesium sulfate is the drug of choice for prevention and treatment of eclamptic seizures. It is superior to diazepam and phenytoin (proven in two RCTs in 1995). The landmark Magpie Trial (>10,000 women, 33 countries) showed magnesium reduced eclamptic seizures by 50% (0.8% vs. 1.9%).
- Regimen: IV bolus loading dose followed by continuous infusion
- Therapeutic serum level: 5-9 mg/dL - slows neuromuscular conduction and depresses CNS irritability
- Duration: Short-term (<48 hours) IV use is recommended by ACOG for seizure prevention/treatment
- Toxicity monitoring (especially in renal impairment):
- Loss of deep tendon reflexes (first sign)
- Flushing, somnolence, muscle weakness
- Decreased respiratory rate (severe toxicity)
- Antidote: Calcium gluconate IV
Note: Long-term use (>5-7 days) as a tocolytic carries FDA warning due to adverse fetal bone effects.
(Brenner and Rector's The Kidney, p. 2157)
4. Corticosteroids
- Fetal lung maturity: Antenatal corticosteroids (betamethasone) are given to accelerate fetal lung maturity when delivery is anticipated before 34 weeks
- HELLP syndrome: High-dose dexamethasone has been used but a recent RCT showed no benefit for routine HELLP management; a subgroup with platelet count < 50,000 may benefit (shorter platelet recovery, shorter hospitalization), but further evidence is needed
5. Management of HELLP Syndrome
HELLP (Hemolysis, Elevated Liver enzymes, Low Platelets) is a severe variant of preeclampsia:
- Clinical course is often rapidly unpredictable - many recommend immediate delivery
- In stable patients at 24-34 weeks with reassuring fetal status, expectant management is an option
- Plasmapheresis (based on pathophysiologic similarity to TTP) has been reported but data are limited and benefit is unclear
6. Prevention Strategies
- Low-dose aspirin (81 mg/day): ACOG recommends starting at 12 weeks in women at high risk (multifetal gestation, prior preeclampsia, chronic hypertension, diabetes, renal disease, autoimmune disease, obesity). Best effect when started before 16 weeks. Evidence in multifetal gestations is less clear.
- Calcium supplementation: May reduce incidence in calcium-deficient populations
- No role for: Bed rest, salt restriction, or routine antioxidants (vitamins C and E)
7. Novel/Emerging Therapies
Research targets the underlying pathophysiology - specifically the excess of sFlt-1 (soluble FMS-like tyrosine kinase-1), an anti-angiogenic factor. Approaches to reduce sFlt-1 (e.g., apheresis) are being explored in pilot studies to allow safer postponement of delivery in early severe preeclampsia.
Quick Summary Table
| Problem | Treatment |
|---|---|
| BP > 150-160/100-110 | Labetalol, Methyldopa, Nifedipine (oral); IV Labetalol or Nicardipine (acute) |
| Seizure prevention/treatment | Magnesium sulfate IV |
| HELLP / severe disease | Deliver; consider dexamethasone if platelets < 50,000 |
| Preterm (<34 wks) delivery anticipated | Betamethasone for fetal lung maturity |
| High-risk prevention | Aspirin 81 mg/day from 12-16 weeks |
| Definitive cure | Delivery |
Sources: Brenner and Rector's The Kidney, 2-Volume Set (Chapter 48, pp. 2156-2161); Creasy & Resnik's Maternal-Fetal Medicine
Explain the pathophysiology of preeclampsia (placental/endothelial dysfunction)
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
I now have comprehensive content and all three key diagrams. Let me compile the full pathophysiology explanation.
Pathophysiology of Preeclampsia: Placental and Endothelial Dysfunction
Preeclampsia is fundamentally a two-stage disease: an early placental stage (poor implantation/ischemia) that triggers a second systemic stage (maternal endothelial dysfunction) responsible for all the clinical manifestations.
Stage 1 - Failure of Spiral Artery Remodeling (The Placental Stage)
In a normal pregnancy, fetal extravillous cytotrophoblasts invade the maternal decidua and the decidual spiral arteries. They destroy the vascular smooth muscle and replace maternal endothelial cells with fetal trophoblastic cells - a process called endovascular invasion. The result is conversion of spiral arteries from narrow, high-resistance vessels into wide, low-resistance, high-capacitance uteroplacental vessels that carry blood at low velocity and pressure, allowing optimal oxygen exchange.
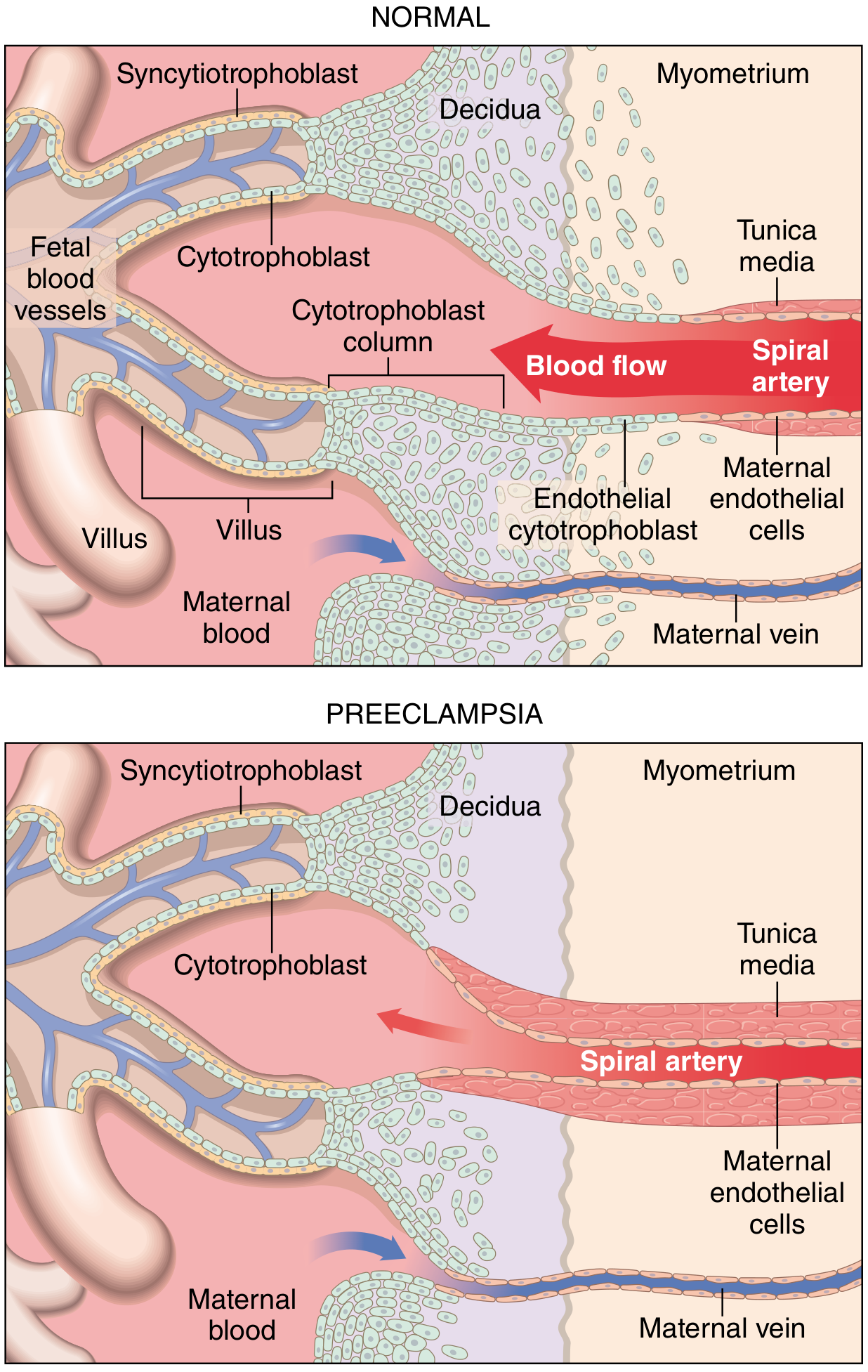
In preeclampsia (bottom), the spiral arteries retain their smooth muscle coat, remain narrow and high-resistance, and trophoblast invasion is shallow - confined to the decidua only, not penetrating the myometrium.
In preeclampsia, this remodeling fails. Trophoblast invasion is abnormally shallow - the spiral arteries retain their smooth muscle coat and remain narrow, high-resistance vessels. Consequences:
- Reduced uteroplacental perfusion
- Placental ischemia and hypoxia - evidenced by expression of hypoxia-inducible factor 1-alpha (HIF-1α) in preeclamptic placentas
- Oxidative stress, NF-κB activation, increased necrotic trophoblast shedding, and proinflammatory interleukin production
- Pathologic inflammation - acute atherosis seen in 10% of preeclamptic placentas (lipid deposition in spiral artery intima)
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 954; Comprehensive Clinical Nephrology 7e, p. 627)
Stage 2 - Angiogenic Imbalance and Systemic Endothelial Dysfunction
The ischemic placenta responds by releasing anti-angiogenic factors into the maternal circulation, causing widespread endothelial injury.
The Two Key Anti-Angiogenic Proteins
1. sFlt-1 (Soluble FMS-like Tyrosine Kinase-1)
- A truncated, soluble form of the VEGF receptor (Flt-1)
- Secreted in massive excess by the syncytiotrophoblast in preeclampsia
- Acts as a "decoy receptor" - circulates freely and traps VEGF and PlGF (placental growth factor), preventing them from binding their endothelial cell-surface receptors
- sFlt-1 levels are detectable in maternal blood before the clinical syndrome appears
2. sEng (Soluble Endoglin)
- A co-receptor for TGF-β1 (transforming growth factor beta-1)
- Excess sEng circulates and traps TGF-β1, blocking its signaling
- TGF-β1 normally stimulates endothelial nitric oxide (NO) production - a potent vasodilator. Blocking it leads directly to systemic vasoconstriction
The diagram below illustrates how these two proteins together cause endothelial dysfunction:
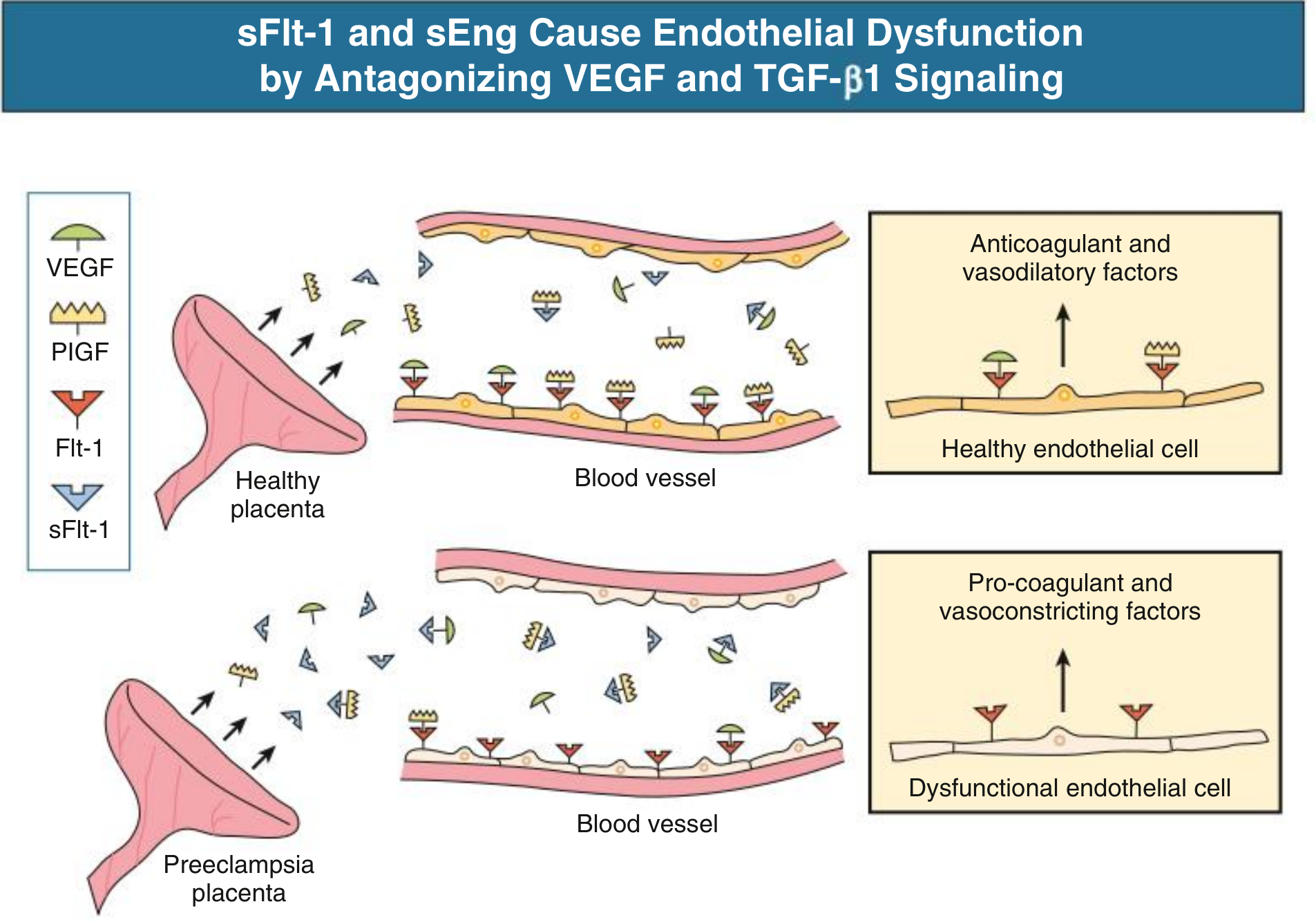
- Healthy placenta (top): VEGF and PlGF bind endothelial Flt-1 receptors → endothelium produces anticoagulant and vasodilatory factors (prostacyclin, nitric oxide)
- Preeclamptic placenta (bottom): Excess sFlt-1 sequesters VEGF/PlGF → endothelium becomes dysfunctional → switches to pro-coagulant and vasoconstrictive state
The net result of sFlt-1 and sEng excess:
- Decreased endothelial prostacyclin (PGI₂) → vasoconstriction and platelet aggregation
- Decreased nitric oxide → systemic vasoconstriction → hypertension
- Decreased angiogenesis → defective placental vascular development (feeds back to worsen placental ischemia)
- Increased vascular permeability → proteinuria and edema
- Pro-coagulant state → thrombosis and HELLP
The full pathogenesis timeline from prepregnancy to postpartum:

(Comprehensive Clinical Nephrology 7e, Fig. 44.4 & 44.5, p. 627; Robbins, p. 954-955)
Organ-Specific Consequences of Endothelial Dysfunction
Kidney - Glomerular Endotheliosis (Hallmark Lesion)
The renal lesion of preeclampsia was first described in 1924 and termed "glomerular endotheliosis" by Spargo et al. It is considered the histological hallmark:
- Marked swelling and vacuolization of glomerular endothelial cells
- Loss of endothelial fenestrae (the openings that allow filtration)
- Fibrinogen/fibrin deposits within and under endothelial cells
- Occlusion of capillary lumina → "bloodless glomerulus" on light microscopy
- Podocyte foot processes are intact early (unlike nephrotic syndrome from other causes)
- Result: ↓ GFR, ↓ renal blood flow (due to increased afferent arteriolar resistance), proteinuria, sodium/water retention
This lesion is caused by VEGF deprivation - podocyte-specific VEGF knockout mice develop identical glomerular endotheliosis and proteinuria. Similarly, cancer patients treated with VEGF antagonists develop renal lesions identical to preeclampsia.
(Brenner and Rector's The Kidney, p. 2150)
Brain - Cerebral Edema and PRES
- Cerebral edema and petechial/parenchymal hemorrhage are hallmarks
- Edema correlates with markers of endothelial damage, not with BP severity - confirming it is endothelial dysfunction-mediated, not purely hypertensive
- MRI pattern: PRES (Posterior Reversible Encephalopathy Syndrome) - vasogenic edema predominantly in parietooccipital subcortical white matter
- Focal hypoperfusion from vasoconstriction → headaches, visual disturbances, cortical blindness, hyperreflexia, seizures
(Brenner and Rector's The Kidney, p. 2155)
Liver
- Irregular subcapsular and intraparenchymal hemorrhages
- Fibrin thrombi in portal capillaries
- Foci of hemorrhagic necrosis
- In HELLP: periportal necrosis, sinusoidal fibrin deposition → elevated transaminases
Placenta (Morphology)
- Placental hypoplasia
- Large, numerous placental infarcts
- Increased syncytial knots (ischemic change)
- Retroplacental hematomas
- Abnormal decidual vessels - fibrinoid necrosis and acute atherosis (lipid deposition in vessel intima)
Coagulation - Thrombosis and HELLP
- Reduced endothelial PGI₂ (prostacyclin - anti-thrombotic, vasodilatory)
- Increased procoagulant factor release
- Thrombosis in arterioles and capillaries throughout placenta, liver, kidneys, brain, and pituitary
- In HELLP: microangiopathic hemolysis, thrombocytopenia, transaminitis
(Robbins, Cotran & Kumar, p. 955)
Summary: The Two-Stage Model
Pre-existing risk factors
(endothelial vulnerability, genetic, immunologic, oxidative stress)
↓
Early pregnancy: Shallow trophoblast invasion
↓
Failure of spiral artery remodeling
↓
Uteroplacental ischemia / Placental hypoxia
↓
Excess sFlt-1 and sEng released from syncytiotrophoblast
↓
Sequestration of VEGF, PlGF, TGF-β1
↓
Systemic maternal endothelial dysfunction
├── ↓ Nitric oxide → Vasoconstriction → Hypertension
├── ↓ Prostacyclin → Pro-thrombotic state → HELLP
├── ↑ Vascular permeability → Proteinuria, Edema
├── Renal: Glomerular endotheliosis → AKI
├── CNS: PRES, hemorrhage, seizures
└── Hepatic: Necrosis, hemorrhage
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 953-956 | Comprehensive Clinical Nephrology 7e, pp. 626-628 | Brenner and Rector's The Kidney, pp. 2149-2157 | Barash Clinical Anesthesia 9e, p. 3508
This is a shared conversation. Sign in to Orris to start your own chat.