Myelodysplastic syndrome
Reading File
Finding Sources
Searching PubMed
"myelodysplastic syndrome"[MeSH Terms] AND treatment
Reading File
Finding Sources
Searching PubMed
"myelodysplastic syndromes"[MeSH Terms] AND "drug therapy"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have excellent comprehensive content from Harrison's 22E (2025), Robbins Pathology, and Goldman-Cecil to compile a thorough response. Let me now compile the complete answer:
Myelodysplastic Syndromes (MDS)
Definition
Myelodysplastic syndromes are a heterogeneous group of clonal hematopoietic stem cell disorders characterized by two core features:
- Cytopenias from ineffective hematopoiesis (the marrow is typically hypercellular yet peripheral blood counts are low - a hallmark paradox)
- High risk of transformation to acute myeloid leukemia (AML)
The bone marrow is partly or wholly replaced by the clonal progeny of a transformed multipotent stem cell that retains the capacity to differentiate into red cells, granulocytes, and platelets - but does so ineffectively and in a disordered manner. MDS is classified as a hematopoietic neoplasm by the WHO, not merely a "pre-leukemic" condition.
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 107
- Robbins & Kumar Basic Pathology, p. 405
Epidemiology
- Incidence: ~15,000 new cases/year in the United States - roughly as common as AML
- Median age at diagnosis: seventh to eighth decade (most patients are 50-80 years old)
- MDS in childhood is rare and implies an underlying genetic disease (e.g., Down syndrome, Fanconi anemia, GATA2 mutations)
- More common in males
- Incidence has been underestimated due to varying diagnostic criteria
Etiology and Risk Factors
Primary (idiopathic): Most cases have no identifiable cause.
Secondary (therapy-related MDS):
- Prior chemotherapy (especially alkylating agents, topoisomerase II inhibitors)
- Prior ionizing radiation therapy
- These therapy-related cases share similar recurrent chromosomal abnormalities with primary MDS
Hereditary predispositions:
- Germline GATA2 mutations (MonoMAC syndrome - susceptibility to viral, mycobacterial, and fungal infections, deficient monocytes, NK cells, B cells)
- Germline RUNX1 mutations (high risk of MDS/leukemia, often preceded by years of thrombocytopenia)
- Fanconi anemia, telomeropathies (TERC, TERT mutations)
- Constitutional SAMD9/SAMD9L mutations
Precursor state: MDS often arises from clonal hematopoiesis of indeterminate prognosis (CHIP) - normal blood counts with clonal "driver" mutations identical to those found in MDS. CHIP progresses to an overt white cell neoplasm at ~1% per year and may also be a cardiovascular disease risk factor.
Pathogenesis
Molecular Mutations (three major categories)
| Category | Examples | Notes |
|---|---|---|
| Epigenetic regulators | TET2, DNMT3A, ASXL1, EZH2, IDH1/IDH2 | DNA methylation & histone modification dysregulation |
| RNA splicing factors | SF3B1, SRSF2, U2AF1, ZRSR2 | Often associated with ring sideroblasts; SF3B1 mutations define a specific subtype |
| Transcription factors | RUNX1, ETV6, TP53 | TP53 loss-of-function (~10% of cases) = complex karyotype, worst prognosis |
Chromosomal Abnormalities
- Monosomies: -5, -7
- Deletions: del(5q), del(7q), del(20q)
- Trisomy: +8
- The 5q- deletion causes heterozygous loss of a ribosomal protein gene (RPS14), which mimics constitutional red cell aplasia - this is the mechanistic basis for lenalidomide sensitivity
Immune Dysregulation
- In lower-risk MDS, an immune pathophysiology may be important - cytopenias can respond to immunosuppressive therapy (as in aplastic anemia)
- The role of the hematopoietic stem cell niche and microenvironment remains incompletely understood
Classification (WHO 5th Edition / ICC 2022)
Both the 2022 WHO 5th Edition and the International Consensus Classification (ICC) now incorporate morphology, cytopenias, bone marrow blast %, cytogenetics, AND molecular findings:
| Subtype | Blasts (BM/PB) | Key Features |
|---|---|---|
| MDS with 5q deletion (MDS-5q) | <5% BM, <2% PB | 5q deletion alone or +1 other abnormality (not -7/7q del); no multi-hit TP53 |
| MDS with SF3B1 mutation (MDS-SF3B1) | <5% BM, <2% PB | SF3B1 mutation; ring sideroblasts; favorable prognosis |
| MDS with low blasts (MDS-LB) | <5% BM, <2% PB | No defining mutation or cytogenetics |
| MDS with excess blasts (MDS-EB) | 5-19% BM or 2-19% PB | Higher AML transformation risk |
| MDS with biallelic TP53 (MDS-biTP53) | Any blast % | Biallelic/multi-hit TP53; very poor prognosis |
| MDS/AML (ICC only) | ≥10% BM | Recognizes disease continuum with AML |
Classic FAB subtypes (RA, RARS, RAEB, RAEB-t, CMML) are now superseded, though familiar to older literature. CMML is now classified as a myelodysplastic/myeloproliferative neoplasm (MDS/MPN).
Clinical Features
Symptoms
- Anemia dominates the early course: fatigue, weakness, dyspnea, pallor
- At least half of patients are asymptomatic at diagnosis - discovered incidentally on CBC
- Bleeding symptoms (due to platelet dysfunction despite adequate counts)
- Increased susceptibility to infection (neutropenia + neutrophil dysfunction)
- Fever and weight loss are more characteristic of myeloproliferative rather than myelodysplastic disease
Physical Examination
- Signs of anemia (pallor, tachycardia)
- ~20% have splenomegaly
- Accompanying autoimmune syndromes occur (Sweet's syndrome, vasculitis)
- VEXAS syndrome should be considered in MDS with coexisting inflammatory disease (caused by somatic UBA1 mutations)
Laboratory Findings
Peripheral Blood
- Anemia (usually macrocytic) - present in most cases
- Bicytopenia or pancytopenia in advanced disease
- Large platelets lacking granules, functionally abnormal
- Pseudo-Pelger-Huet neutrophils (hyposegmented nuclei), hypogranulated neutrophils, Döhle bodies
- Circulating myeloblasts (correlates with marrow blast %)
- WBC usually normal or low (except CMML where monocytosis occurs)
- Associated PNH clone may be present
Bone Marrow
- Usually normocellular or hypercellular (paradoxically, despite peripheral cytopenias)
- ~20% are hypocellular - can be confused with aplastic anemia
- Dysplastic changes in all three lineages (see morphology below)
- Myeloblasts <20% by definition (≥20% = AML)
Bone Marrow Morphology
The image below from Robbins Pathology illustrates classic dysplastic changes in MDS:
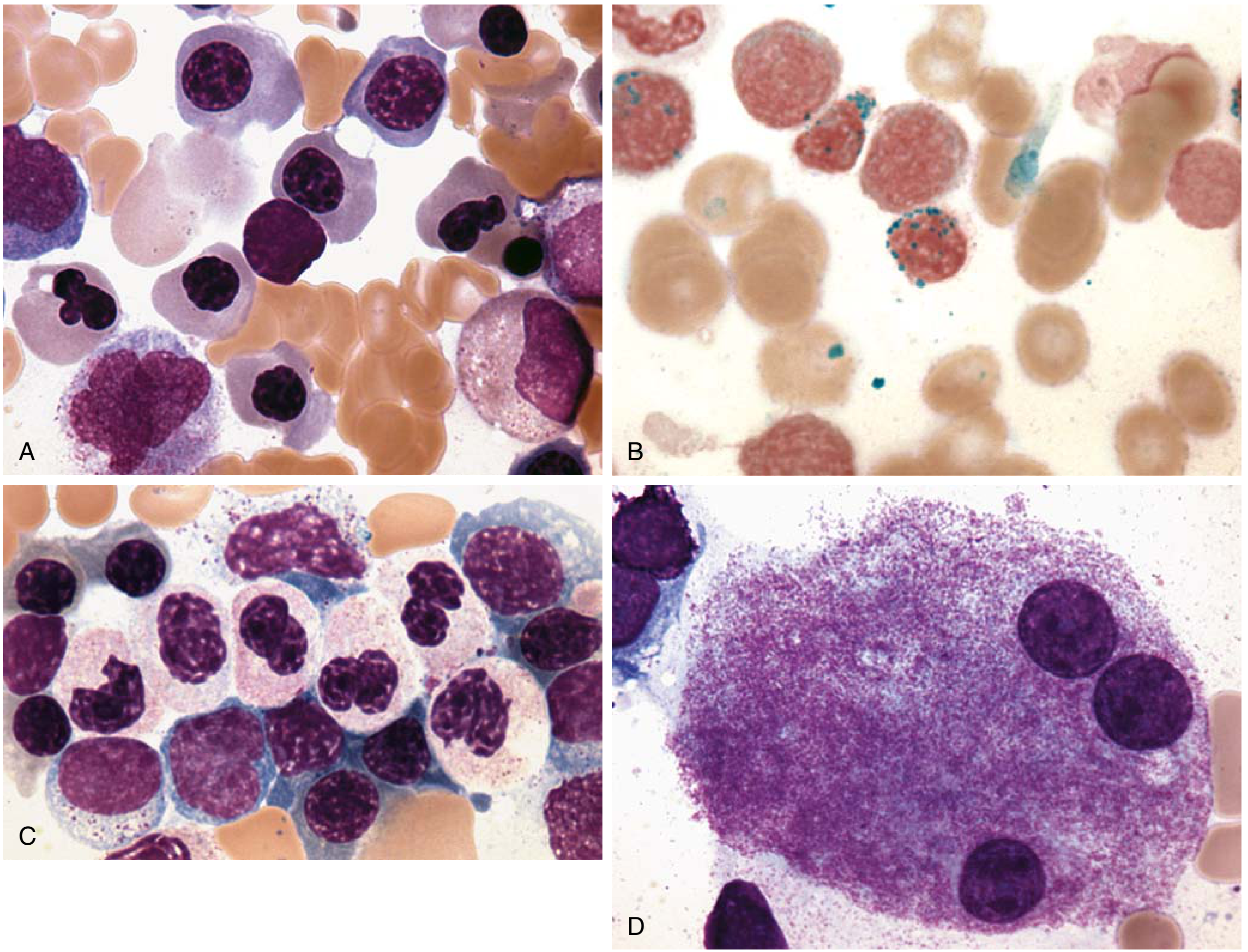
(A) Erythroid progenitors with abnormal irregular nuclei (megaloblastoid change, nuclear budding, karyorrhexis)
(B) Ring sideroblasts - iron-laden mitochondria encircling ≥1/3 of nucleus (Prussian blue stain)
(C) Abnormal neutrophils - hyposegmented nuclei (pseudo-Pelger-Huet), hypogranulation
(D) Abnormal megakaryocyte with three separate nuclei (nuclear hyperlobation or hypolobation)
Cytogenetics & Molecular Testing
- Conventional karyotype: abnormal in ~50% of primary MDS, ~80% of therapy-related MDS
- Next-generation sequencing is now routinely performed - detects mutations in >90% of MDS cases
- Testing for SF3B1, SRSF2, TET2, ASXL1, DNMT3A, TP53, RUNX1, etc.
Prognosis: IPSS-R Scoring
The Revised International Prognostic Scoring System (IPSS-R) is the standard tool for risk stratification. It incorporates:
| Variable | Factors |
|---|---|
| Cytogenetics | Very good / Good / Intermediate / Poor / Very poor |
| Bone marrow blast % | ≤2% / >2-<5% / 5-10% / >10% |
| Hemoglobin | ≥10 / 8-<10 / <8 g/dL |
| Platelets | ≥100 / 50-<100 / <50 ×10⁹/L |
| Neutrophils | ≥0.8 / <0.8 ×10⁹/L |
Risk categories: Very Low / Low / Intermediate / High / Very High
- Median overall survival: 9-29 months (varies by risk group)
- AML transformation: 10-40% of patients
- Molecular IPSS (IPSS-M) incorporating mutation data now provides better prognostic discrimination
Treatment
Supportive Care (all risk groups)
- Red cell transfusions for symptomatic anemia
- Platelet transfusions for significant bleeding
- Iron chelation (deferoxamine, deferasirox) for transfusion-dependent patients with iron overload
- G-CSF for severe neutropenia with infections (typically not routinely given)
- Growth factors: Erythropoiesis-stimulating agents (ESAs - epoetin alfa, darbepoetin) for patients with low endogenous EPO levels; response rate ~25-40%
Lower-Risk MDS (IPSS-R Very Low/Low/Intermediate)
Lenalidomide (for del 5q):
- Highly effective in MDS with 5q- syndrome
- A high proportion become transfusion-independent with near-normal hemoglobin
- Cytogenetics normalize in many patients
- Administered orally; toxicities include myelosuppression and DVT/PE risk
Immunosuppression (younger patients with lower-risk MDS):
- Antithymocyte globulin (ATG) + cyclosporine
- Anti-CD52 monoclonal antibody alemtuzumab
- Especially effective in patients <60 years with HLA-DR15 positivity and a PNH clone
Imetelstat (telomerase inhibitor):
- FDA-approved for ESA-relapsed/refractory lower-risk MDS
- Phase 3 IMerge trial (Lancet, 2024) demonstrated transfusion independence vs placebo [PMID 38048786]
Higher-Risk MDS (IPSS-R High/Very High)
Hypomethylating agents (HMAs) - first-line:
- Azacitidine (5-azacytidine): subcutaneous, daily ×7 days every 28 days; improves blood counts and survival vs best supportive care; response in ~50% of patients; FDA-approved for all MDS subtypes; responses are dependent on continued administration
- Decitabine: IV, various schedules (3-10 days); response in 30-50%, duration ~1 year
- Oral decitabine-cedazuridine (Inqovi): FDA-approved oral formulation; Phase 3 ASCERTAIN trial (Lancet Haematol, 2024) showed non-inferiority to IV decitabine [PMID 38135371]
Major toxicity of HMAs: myelosuppression (worsening cytopenias, especially early in treatment)
Venetoclax + azacitidine (emerging):
- Phase 1/2 data from Garcia et al. (Blood, 2025) show promising activity for treatment-naive high-risk MDS [PMID 39652823]
- Not yet standard of care but incorporated into clinical trials
Curative Intent: Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT)
- The only known curative treatment for MDS
- Survival ~50% at 3 years in selected cohorts (improving with better supportive care and donor matching)
- Matched unrelated donor results are similar to sibling donor results
- Barriers: Advanced age of most patients, comorbidities, donor availability, patient preferences
- Only a small proportion of MDS patients ultimately undergo transplantation
- Reduced-intensity conditioning (RIC) now extends eligibility to older patients (though higher relapse risk)
- Transplant timing dilemma: high-risk patients most need it but tolerate it least; low-risk patients tolerate it best but may do well for years with less aggressive therapy
- Pre-transplant HMA therapy (bridging) is common; prior therapy does not appear to significantly harm outcomes
MDS vs. AML Distinction
- AML: ≥20% myeloblasts in BM or peripheral blood
- MDS with excess blasts: 5-19% BM blasts (high leukemic transformation risk)
- The ICC (2022) introduced MDS/AML (≥10% blasts) to recognize the biological continuum - these cases often have similar outcomes regardless of which side of the 20% threshold they fall on
Key Associations to Remember
| Feature | Significance |
|---|---|
| Ring sideroblasts + SF3B1 mutation | MDS-SF3B1 subtype; relatively favorable prognosis |
| del(5q) alone | MDS-5q; lenalidomide-sensitive; favorable prognosis in women |
| Monosomy 7 / del(7q) | Poor prognosis |
| Complex karyotype (≥3 abnormalities) | Poor prognosis |
| Multi-hit TP53 | Worst prognosis; separate classification category |
| Hypocellular MDS | Can mimic aplastic anemia; responds to immunosuppression |
| VEXAS syndrome | MDS + refractory inflammatory disease; somatic UBA1 mutations |
| CHIP | Asymptomatic precursor to MDS; ~1%/year progression |
Recent Developments (2024-2026)
- Oral decitabine-cedazuridine (Inqovi) confirmed non-inferior to IV decitabine in Phase 3 ASCERTAIN trial - improves convenience significantly [PMID 38135371]
- Imetelstat (telomerase inhibitor) Phase 3 trial confirmed efficacy in lower-risk, ESA-refractory MDS with transfusion-independence benefit [PMID 38048786]
- Venetoclax + azacitidine showing promising Phase 1/2 data for high-risk MDS (Blood, 2025) [PMID 39652823]
- Both WHO 5th Edition (2022) and ICC (2022) now use molecular markers to define subtypes - the era of purely morphologic classification is ending
- IPSS-M (molecular IPSS) provides refined prognostication incorporating gene mutation data
Sources: Harrison's Principles of Internal Medicine 22E (2025), Ch. 107; Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine; Robbins, Cotran & Kumar Pathologic Basis of Disease
Thalassemia
Reading File
Finding Sources
Searching PubMed
"thalassemia"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive content from Harrison's 22E (2025) and Robbins. Let me compile the full response:
Thalassemia
Definition
Thalassemia is a group of inherited disorders caused by mutations that reduce the synthesis of either α-globin or β-globin chains, leading to an imbalance in globin chain production. The hallmark is unbalanced globin synthesis: the excess unpaired normal chain precipitates inside developing erythroblasts, causing membrane damage, ineffective erythropoiesis, and hemolysis. The result is anemia from both reduced red cell production and reduced red cell lifespan.
- The name comes from the Greek thalassa ("sea"), reflecting the original description in Mediterranean populations.
- ~500 unique thalassemia-causing mutations are known; clinically, β-thalassemia and α-thalassemia are the most important.
Genetics and Normal Hemoglobin Structure
Adult hemoglobin A (HbA) = α₂β₂ tetramer.
| Globin chain | Gene locus | Copies per diploid cell |
|---|---|---|
| α-globin | Chromosome 16 | 4 (2 per haplotype) |
| β-globin | Chromosome 11 | 2 (1 per haplotype) |
Other hemoglobins:
- HbF (fetal) = α₂γ₂ - elevated in thalassemia, partially compensatory
- HbA₂ = α₂δ₂ - elevated in β-thalassemia trait (diagnostic marker)
- HbH = β₄ tetramers - form in α-thalassemia when α chains are severely reduced
- Hb Bart's = γ₄ tetramers - form in severe α-thalassemia in fetal life
Epidemiology
- Estimates: 1-5% of the world's population carries a thalassemia mutation; in some locales, most people are carriers
- Common in regions where Plasmodium falciparum malaria was (is) endemic: the Mediterranean basin, Middle East, tropical Africa, Indian subcontinent, Southeast Asia
- Evolutionary explanation: Heterozygous carriers are protected from severe P. falciparum malaria - this drove the mutation to polymorphic frequencies
- ~40,000 β-thalassemia patients are born yearly worldwide
- About 30% of African Americans carry the common -α³·⁷ chromosome (single-gene α-thalassemia)
- HbH disease and Hb Bart's hydrops fetalis are most common in southern China and Southeast Asia
Part 1: β-Thalassemia
Molecular Pathogenesis
β-thalassemia is caused by point mutations (rarely deletions) in the β-globin gene. Over 100 causative mutations are known, falling into three classes:
| Mutation Type | Mechanism | Severity |
|---|---|---|
| Splicing mutations (most common) | Destroy normal RNA splice junctions OR create ectopic splice sites; some allow partial normal splicing | β⁰ or β⁺ |
| Promoter region mutations | Reduce transcription by 75-80%; some normal β-globin still made | β⁺ |
| Chain terminator mutations (most common cause of β⁰) | Nonsense mutations or frameshift insertions/deletions; no functional β-globin produced | β⁰ |
- β⁰ mutations: Completely prevent β-globin accumulation
- β⁺ mutations: Cause reduced (but detectable) β-globin synthesis
Downstream pathophysiology of β-chain deficit:
- Excess α-chains accumulate and precipitate within erythroblasts → membrane lipid oxidation and damage
- Ineffective erythropoiesis (intramedullary destruction of erythroid precursors) - the predominant cause of anemia
- Reduced deformability and phosphatidylserine exposure → extravascular and intravascular hemolysis of circulating cells
- Severe anemia → compensatory erythroid marrow expansion → skeletal deformities
- Increased intestinal iron absorption + transfusional iron → iron overload in liver, heart, and endocrine organs
Classification of β-Thalassemia
| Clinical Syndrome | Genotype | Hb (g/dL) / MCV (fL) | HbA₂ | HbF | Clinical Features |
|---|---|---|---|---|---|
| β-Thalassemia trait (minor) | Heterozygous (β⁺/β or β⁰/β) | 10-14 / 60-80 | 4-6% (elevated - diagnostic) | 1-2% | Asymptomatic; mild/no anemia; microcytic hypochromic RBCs |
| Non-transfusion-dependent (intermedia) | Variable compound heterozygous | 7-12 / 65-80 | Elevated | 10-40% | Moderate anemia; regular transfusions NOT required; significant iron loading from gut |
| Transfusion-dependent (major) | Homozygous β⁰/β⁰ or compound heterozygous | <7 / <65 | Variable | >60-90% | Severe anemia; regular transfusions required lifelong |
Clinical Features of β-Thalassemia Major (Cooley's Anemia)
If inadequately treated:
- Severe hemolytic anemia - jaundice, pallor, fatigue from early childhood
- Massive hepatosplenomegaly - extramedullary hematopoiesis in liver, spleen
- Skeletal deformities from marrow expansion:
- "Chipmunk" facies (maxillary hypertrophy)
- Frontal bossing
- "Hair-on-end" appearance on skull X-ray (classic radiographic finding)
- Pathological fractures
- Growth retardation, delayed puberty
- Iron overload complications (even before transfusions, from increased gut absorption):
- Cardiomyopathy (leading cause of death)
- Cirrhosis, hepatic failure
- Endocrinopathies: diabetes, hypogonadism, hypothyroidism, hypoparathyroidism, adrenal insufficiency
- Osteoporosis
With adequate treatment (transfusion + iron chelation), most complications are preventable and life expectancy extends to at least 50 years.
Blood Film in Thalassemia Intermedia
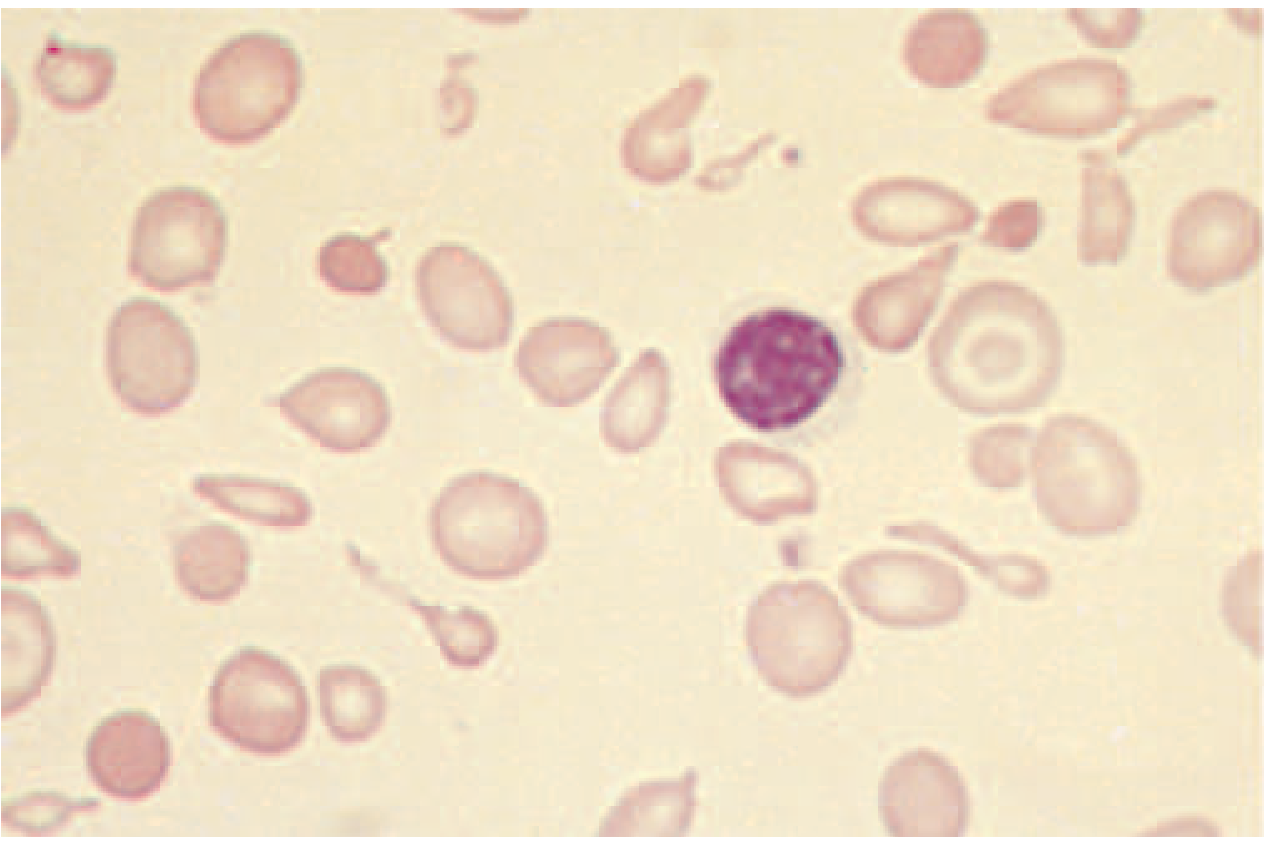
Target cells, marked anisocytosis and poikilocytosis, hypochromia, and microcytosis. Nucleated red blood cells may be seen in more severe forms.
Diagnosis of β-Thalassemia
- CBC: Microcytic (low MCV), hypochromic anemia; normal/low reticulocytes (ineffective erythropoiesis); anisocytosis, poikilocytosis, target cells, basophilic stippling
- Hemoglobin HPLC/electrophoresis:
- Trait: HbA₂ elevated (4-6%) - the diagnostic hallmark
- Major: predominantly HbF (>60%), markedly reduced or absent HbA, elevated HbA₂
- Serum ferritin and iron studies: Iron overload (NOT deficiency - do not give iron!)
- Bone marrow: Erythroid hyperplasia, ineffective erythropoiesis
- DNA analysis: Required before genetic counseling and antenatal diagnosis to identify the specific mutation
- Must exclude iron deficiency (which also causes microcytosis but with low ferritin, low HbA₂)
Part 2: α-Thalassemia
Molecular Pathogenesis
- α-thalassemia is most often caused by deletions of α-globin genes (rather than point mutations)
- Each chromosome 16 has two α-globin genes → diploid individuals have 4 α-globin genes total
- Severity is proportional to the number of deleted/non-functional α-globin genes
Classification of α-Thalassemia
| Syndrome | Functional α genes | Genotype | Hb (g/dL) / MCV | Clinical Features |
|---|---|---|---|---|
| Silent carrier | 3 of 4 | -α/αα | Normal (~normal) | Completely asymptomatic; slight microcytosis only |
| α-Thalassemia trait (minor) | 2 of 4 | --/αα (Asian) or -α/-α (African/Asian) | 12-15 / 65-80 | Asymptomatic; resembles β-thalassemia minor; normal HbA₂ (unlike β-thal trait) |
| HbH disease | 1 of 4 | --/-α | 5-12 / 60-70 | Moderate-severe anemia; HbH (β₄) tetramers form; Hb Bart's at birth |
| Hb Bart's hydrops fetalis (α-thalassemia major) | 0 of 4 | --/-- | Lethal | Predominantly Hb Bart's (γ₄); fatal in utero or shortly after birth |
Asian vs. African distinction:
- Asians: commonly carry the --/αα haplotype (both deletions on one chromosome) → at risk for HbH disease and hydrops in offspring
- Africans: commonly carry -α/-α (one deletion per chromosome) → very rarely produce severely affected offspring even when both parents are carriers
Clinical Features of HbH Disease
- Moderately severe hemolytic anemia (resembles β-thalassemia intermedia)
- HbH (β₄ tetramers) has extremely high O₂ affinity → delivers little O₂ to tissues → disproportionate tissue hypoxia
- HbH oxidizes and precipitates → intracellular inclusions → spleen sequestration
- Hepatosplenomegaly, jaundice, thalassemic facies, and growth impairment in 20-50% of cases
- Iron loading occurs but is less severe than in β-thalassemia
- Diagnosis: HbH inclusions on brilliant cresyl blue stain; HPLC shows traces to 40% HbH in adults
Clinical Features of Hb Bart's Hydrops Fetalis
- Deletion of all 4 α-globin genes → no functional hemoglobin can form
- Hb Bart's (γ₄) has extremely high O₂ affinity - unable to deliver O₂ to fetal tissues
- Third trimester fetal distress → severe pallor, generalized edema (hydrops), massive hepatosplenomegaly, ascites
- Without intrauterine transfusion: fatal in utero or shortly after birth
- With intrauterine transfusion + perinatal intensive care: survival possible, but ~40% have growth retardation, ~20% neurodevelopmental delay
- Maternal complications: preeclampsia, polyhydramnios, antepartum hemorrhage, difficult delivery
- Prevention (antenatal screening + diagnosis) is the best approach
Part 3: Treatment
Supportive Care
Regular blood transfusions (transfusion-dependent thalassemia):
- Every 2-4 weeks with goal pretransfusion Hb of 9-10.5 g/dL
- Suppresses ineffective erythropoiesis, prevents skeletal deformities
- Must be started early and continued lifelong without interruption
- Use leukodepleted, antigen-matched (to minimize alloimmunization) packed red cells
Iron chelation (mandatory with transfusion therapy):
- Prevents accumulation of toxic iron in heart, liver, and endocrine organs
- Indicated when ferritin is consistently elevated (generally >1,000-2,500 mcg/L)
- Three chelating agents available:
| Drug | Route | Notes |
|---|---|---|
| Deferoxamine | IV/SC (8-12 hr infusion) | Oldest; most experience; cumbersome |
| Deferasirox (Exjade/Jadenu) | Oral once daily | First-line oral agent; GI side effects, renal monitoring needed |
| Deferiprone | Oral three times daily | Particularly effective for cardiac iron; risk of agranulocytosis |
- A Cochrane systematic review (2023) examined calcium channel blockers for preventing thalassemia cardiomyopathy alongside iron chelation [PMID 37975597]
Improving Ineffective Erythropoiesis
Luspatercept (Reblozyl):
- Fusion protein (activin type IIB receptor extracellular domain + IgG Fc)
- Binds TGF-β superfamily ligands → reduces Smad2/3 signaling → enhances late-stage erythropoiesis
- Given subcutaneously 1 mg/kg every 3 weeks
- Phase 3 BELIEVE trial: 33% reduction in transfusion requirements in transfusion-dependent β-thalassemia
- FDA-approved for adults with transfusion-dependent β-thalassemia
Hydroxyurea:
- Increases HbF synthesis; used in some non-transfusion-dependent cases
- More effective in sickle cell disease but used as adjunctive therapy in some thalassemia patients
Splenectomy:
- Reduces hypersplenism and transfusion requirements in selected patients
- Increased risk of overwhelming post-splenectomy infection and thrombosis - performed less frequently now
Curative Treatments
Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT):
- The only established curative treatment
- Best results in young children (<7 years), without organ damage from iron overload
- Class I patients (no hepatomegaly, no fibrosis, adequate chelation): >90% event-free survival
- Matched unrelated donor transplants now yield outcomes similar to sibling donors
- HbH disease: transfusions not usually needed; allo-HSCT rarely indicated
Gene Therapy (approved 2022-2023):
- CRISPR/Cas9 - Casgevy (exagamglogene autotemcel): Edits the BCL11A gene (erythroid enhancer) to reactivate γ-globin production → induces HbF
-
90% of 52 patients aged 12-35 with transfusion-dependent β-thalassemia achieved transfusion independence with total Hb ≥12 g/dL and HbF ≥10 g/dL
-
- Lentiviral gene therapy - Zynteglo (betibeglogene autotemcel): Adds a functional HbA^T87Q^ β-globin gene; best results in non-β⁰/β⁰ genotypes
- A 2026 systematic review of CRISPR trials confirmed sustained responses in thalassemia and sickle cell disease [PMID 39794549]
Screening, Counseling, and Antenatal Diagnosis
- Carrier screening: CBC with red cell indices (microcytosis + normal iron → suspect thalassemia trait) → HbA₂ quantification → DNA mutation analysis
- Genetic counseling: Essential before conception or early in pregnancy when both partners are carriers
- Antenatal diagnosis: Chorionic villus sampling (10-12 weeks) or amniocentesis for DNA analysis; can identify β⁰/β⁰ or --/-- fetuses
- Effective screening programs have substantially reduced new case births in Italy, Cyprus, and other high-prevalence regions
Key Distinguishing Features: β- vs. α-Thalassemia Trait
| Feature | β-Thalassemia Trait | α-Thalassemia Trait |
|---|---|---|
| Mechanism | Point mutations in β-globin gene | Gene deletions in α-globin locus |
| HbA₂ | Elevated (4-6%) | Normal or low |
| HbF | Slightly elevated (1-3%) | Normal |
| MCV | Low (<80 fL) | Low (<80 fL) |
| Anemia | Mild or absent | Mild or absent |
| Diagnosis confirmation | HPLC + DNA | HPLC (HbH inclusions) + DNA |
Important: Never treat microcytic anemia due to thalassemia trait with iron supplements - iron stores are normal or elevated. Always exclude iron deficiency first with ferritin/serum iron.
Summary: Clinical Spectrum at a Glance
| Condition | Gene Status | Main Problem | Key Treatment |
|---|---|---|---|
| β-thal trait | Heterozygous β⁺ or β⁰ | None (carrier state) | Counseling only |
| β-thal intermedia | Compound heterozygous | Moderate anemia ± iron loading | Situational transfusions; luspatercept; chelation |
| β-thal major | Homozygous β⁰/β⁰ | Severe anemia, iron overload | Regular transfusions + iron chelation; HSCT/gene therapy |
| α-thal silent carrier | 3/4 genes | None | None |
| α-thal trait | 2/4 genes | Mild microcytosis | Counseling; avoid iron |
| HbH disease | 1/4 genes | Moderate hemolytic anemia | Situational transfusions; avoid oxidant drugs |
| Hb Bart's hydrops | 0/4 genes | Fatal fetal hydrops | Intrauterine transfusion; HSCT post-birth; prevention |
Sources: Harrison's Principles of Internal Medicine 22E (2025), Ch. 103; Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine, Ch. 165
Hb electrophoresis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Finding Sources
I now have excellent visual and textual material from Tietz, Quick Compendium, and Harriet Lane. Let me compile the comprehensive answer:
Hemoglobin Electrophoresis
Purpose
Hemoglobin (Hb) electrophoresis is the separation and identification of hemoglobin variants based on differences in molecular charge and size. It is used to:
- Diagnose hemoglobinopathies (sickle cell disease, HbC, HbE, HbD, etc.)
- Diagnose thalassemias (especially β-thalassemia trait via elevated HbA₂)
- Newborn screening for clinically significant Hb disorders
- Confirm sickle cell preparations (all positive sickle tests must be confirmed by electrophoresis or isoelectric focusing)
Normal Hemoglobin Components
| Hemoglobin | Structure | Normal Adult % | Normal Newborn % |
|---|---|---|---|
| HbA | α₂β₂ | >96-97% | 10-40% |
| HbA₂ | α₂δ₂ | 1.5-3.5% (up to 3%) | <1% |
| HbF | α₂γ₂ | <1% (adults) | 60-90% at birth |
Normal adult pattern: >97% HbA, <3% HbA₂, <1% HbF, no other bands.
Methods
1. Alkaline Electrophoresis (pH 8.4-8.6) - Cellulose Acetate
The traditional first-line method. Hemoglobins migrate toward the anode (positive electrode) at alkaline pH. The more negatively charged the molecule, the faster it migrates.
Migration order (fastest → slowest, cathode to anode):
← Cathode (-) ←————————————————————————————→ Anode (+)
C A₂ E O S D G F A Bart's H
|___|___|___| |___|___| |___| |_________|
(C position) (S position) (F pos.) (fast Hbs)
Key migration positions (alkaline pH 8.6):
| Position | Hemoglobins that co-migrate |
|---|---|
| C zone (slowest) | HbC, HbA₂, HbE, HbO-Arab |
| S zone | HbS, HbD-Punjab, HbG-Philadelphia, Hb-Lepore |
| F zone | HbF |
| A zone (major) | HbA (normal) |
| Fast zone (fastest) | HbH (β₄), Hb Bart's (γ₄) |
Critical limitation: Cannot distinguish HbS from HbD, HbG, or Hb-Lepore (all migrate to the S position). Acid electrophoresis is required to differentiate.
2. Acid Electrophoresis (pH 6.0-6.2) - Citrate Agar
The complementary second-line method used to resolve ambiguous alkaline results. At acid pH, migration patterns differ due to different interactions with the agar matrix.
Migration positions (acid pH 6.2) - key distinctions from alkaline:
| Hemoglobin | Alkaline position | Acid position | Interpretation |
|---|---|---|---|
| HbS | S | S | Stays at S - confirmed |
| HbD | S (same as HbS!) | A | Separates from HbS |
| HbG | S (same as HbS!) | A | Separates from HbS |
| Hb-Lepore | S (same as HbS!) | A | Separates from HbS |
| HbC | C | C | Stays at C |
| HbE | C (same as HbC!) | A (migrates to A) | Separates from HbC |
| HbO-Arab | C (same as HbC!) | C | Stays with HbC |
| HbF | F | F | Same position |
| HbA | A | A | Same position |
Mnemonic for acid gel: "AFSC" - HbA, HbF, HbS, HbC are the four major bands visible; variants that co-migrated with S at alkaline pH now move toward A.
The electrophoresis gel image below (Tietz Textbook) shows alkaline (left) and acid (right) patterns for various hemoglobinopathies:
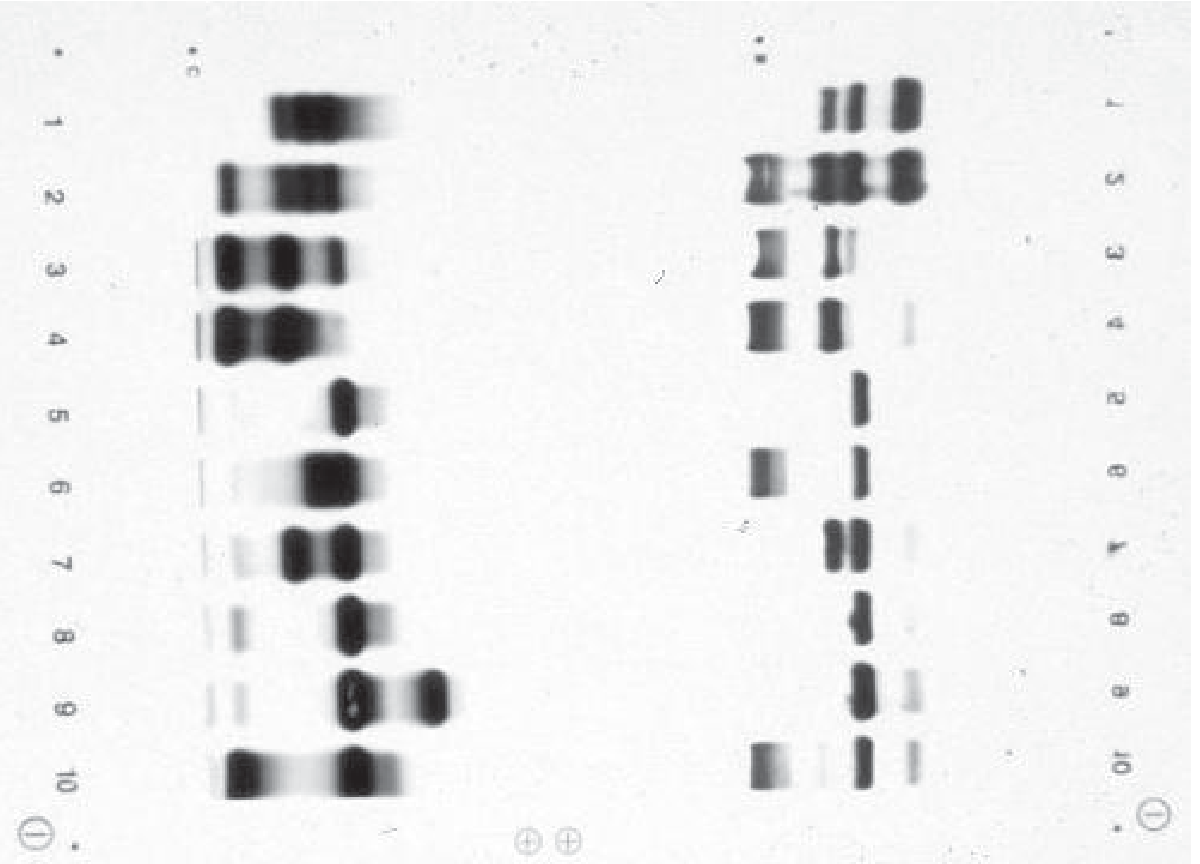
Note: Lane 8 (β-thalassemia trait) shows the characteristic elevated HbA₂ band at the C zone on alkaline electrophoresis.
3. High Performance Liquid Chromatography (HPLC) - Current Gold Standard
HPLC is now the preferred method for hemoglobin analysis. Individual molecules elute from an ion-exchange column at different characteristic retention times, allowing identification and accurate quantification.
Advantages over electrophoresis:
- Accurately quantifies HbA₂ and HbF (electrophoresis cannot do this reliably)
- Higher throughput and automation
- Also quantifies HbA1c (glycated hemoglobin for diabetes monitoring)
- Better sensitivity for small peaks
Normal HPLC chromatogram (adult):
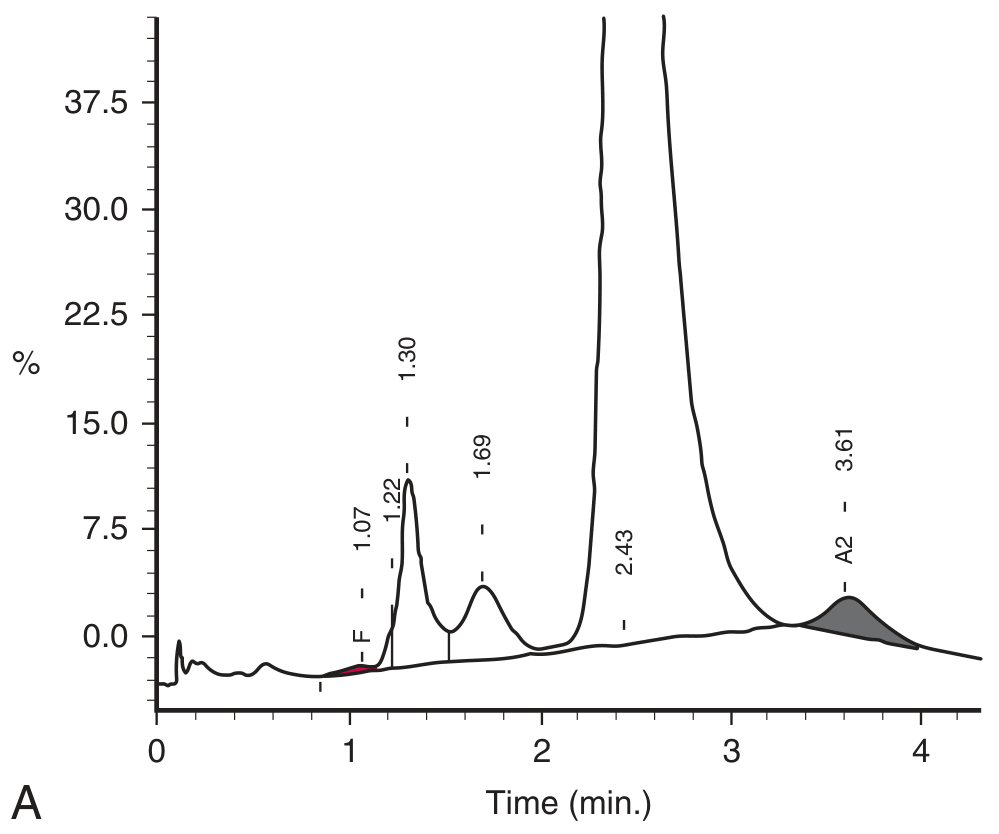
HPLC chromatogram in β-thalassemia trait:
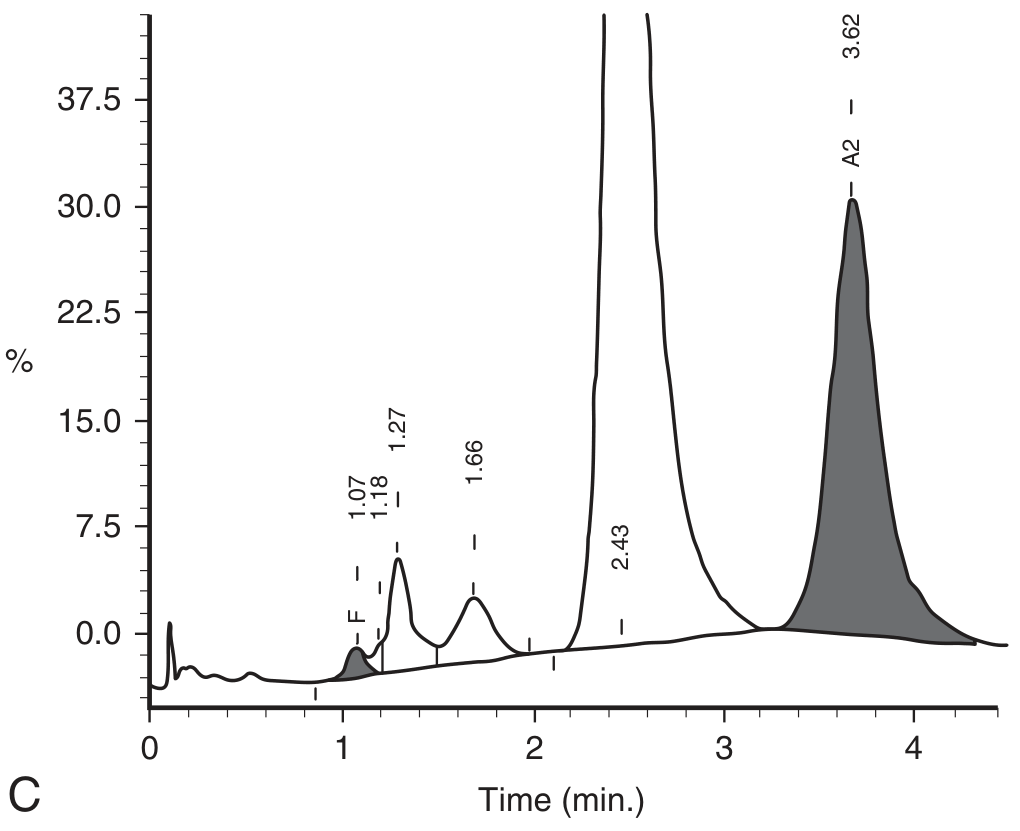
The markedly elevated HbA₂ peak (shaded gray) is the diagnostic signature of β-thalassemia trait.
Key limitations of HPLC:
- HbE and HbA₂ co-elute (similar retention times) - cannot easily separate
- HbC and HbO-Arab co-elute - cannot easily separate
- Capillary zone electrophoresis resolves HbE from HbA₂ (important for accurate HbA₂ quantification in populations where HbE is common)
- Does NOT help separate HbD from HbG and Hb-Lepore
4. Isoelectric Focusing (IEF)
- Separates Hb variants by their isoelectric point (pI)
- Widely used in newborn screening programs (more sensitive than electrophoresis for detecting HbF fractions)
- Part of mandatory newborn screening in many U.S. states
5. Capillary Zone Electrophoresis (CZE)
- Separates Hb variants in a capillary tube at alkaline pH
- Can accurately quantify HbA₂ even in the presence of HbE (unlike HPLC)
- Increasingly used as a standalone confirmatory method
Interpreting Hb Electrophoresis Results
Normal Adult Pattern
| Fraction | Value |
|---|---|
| HbA | >96% |
| HbA₂ | 1.5-3.5% |
| HbF | <1% |
| Abnormal bands | None |
β-Thalassemia Trait (Minor)
| Fraction | Value |
|---|---|
| HbA | ~93-95% (reduced) |
| HbA₂ | ≥4% (elevated - DIAGNOSTIC) |
| HbF | Normal to slightly elevated (1-3% in 30-40% of cases) |
| Abnormal bands | None |
- HbA₂ ≥4% is the diagnostic hallmark of β-thalassemia trait
- Borderline 3.1-3.9%: consider heterozygous KLF1 mutations or concomitant δ-globin mutation
- Must be quantified by HPLC (densitometric scanning of gel is inaccurate)
- False causes of elevated HbA₂: megaloblastic anemia, hyperthyroidism, some unstable Hbs, HIV in women
β-Thalassemia Major
| Fraction | Value |
|---|---|
| HbA | Absent or severely reduced (<10%) in β⁰/β⁰ |
| HbF | Markedly elevated (>60-90%) - compensatory |
| HbA₂ | Variable (may be elevated but can be misleadingly "normal" due to dilution by HbF) |
α-Thalassemia Trait
| Fraction | Value |
|---|---|
| HbA | Normal |
| HbA₂ | Normal or LOW (key distinction from β-thal trait) |
| HbF | Normal |
| Abnormal bands | None |
- Electrophoresis is typically normal in α-thalassemia trait
- Diagnosis is confirmed by DNA analysis (gene deletion testing)
Sickle Cell Trait (HbAS)
| Fraction | Value |
|---|---|
| HbA | ~55-60% (majority) |
| HbS | ~35-45% |
| HbA₂ | Normal (~2%) |
| HbF | Normal |
- Band in S position on alkaline electrophoresis → confirm with acid gel (stays at S)
- Sickle screen (sodium metabisulfite / Sickledex) must be confirmed by electrophoresis
- More HbA than HbS (unlike sickle cell disease where no HbA is present)
Sickle Cell Disease (HbSS)
| Fraction | Value |
|---|---|
| HbA | Absent (no normal β-globin) |
| HbS | ~80-90% (dominant band) |
| HbF | Elevated (1-20%; variable - patients with high HbF have milder disease) |
| HbA₂ | ~2-3.5% |
- Single S band at alkaline pH; confirmed at S position on acid gel
- Sickle screen positive
HbSC Disease
| Fraction | Value |
|---|---|
| HbA | Absent |
| HbS | ~45-50% |
| HbC | ~45-50% |
| HbF | Variable |
- Two bands: one at S position, one at C position (alkaline gel)
- Acid gel: S stays at S; C stays at C - confirms both
- Clinical severity intermediate between sickle trait and HbSS
HbS/β⁰-Thalassemia
| Fraction | Value |
|---|---|
| HbA | Absent (no functional β-globin from either allele) |
| HbS | ~80-90% |
| HbF | Elevated |
| HbA₂ | Elevated (≥4%) |
- Clinically indistinguishable from HbSS
- Key distinguishing feature: elevated HbA₂ (not seen in HbSS)
- Newborn screen: FS pattern
HbS/β⁺-Thalassemia
| Fraction | Value |
|---|---|
| HbA | Present but reduced (5-30%) |
| HbS | ~60-75% |
| HbF | Elevated |
| HbA₂ | Elevated (≥4%) |
- HbA IS present (unlike HbSS and HbS/β⁰), but HbS > HbA
- Milder clinical course than HbSS
- Newborn screen: FSA pattern
HbC Disease (HbCC)
| Fraction | Value |
|---|---|
| HbA | Absent |
| HbC | >90% |
| HbF | Variable |
- HbC = β⁶Glu→Lys (vs HbS = β⁶Glu→Val, same position, different amino acid)
- Alkaline gel: band at C position; acid gel: stays at C position
- Mild hemolytic anemia; numerous target cells on blood smear
- HbC trait (AC): ~60% HbA + ~40% HbC; usually asymptomatic
HbE Trait (AE)
| Fraction | Value |
|---|---|
| HbA | ~65-70% |
| HbE | ~25-30% |
| HbA₂ | Elevated (HbE co-elutes with HbA₂ on HPLC) |
- Alkaline gel: band at C position (same as HbC)
- Acid gel: band at A position (migrates away from HbC - distinguishes E from C)
- HbE/β-thalassemia: one of the most common severe hemoglobin disorders worldwide (Southeast Asia)
- Capillary electrophoresis needed to separate HbE from HbA₂ for accurate quantification
Neonatal Hemoglobin Electrophoresis Patterns
Hemoglobin variants are reported in order of decreasing abundance (e.g., FA = more HbF than HbA):
| Pattern | Interpretation |
|---|---|
| FA | Normal newborn (HbF predominant, HbA present) |
| FAS | Sickle cell trait (benign) |
| FS | Sickle cell disease (SS) or S/β⁰-thalassemia - clinically significant |
| FSA | S/β⁺-thalassemia - clinically significant sickling disorder |
| FSC | HbSC disease - clinically significant |
| FAC | HbC trait (benign) |
| FC | Homozygous HbC disease (mild disorder) |
| F only | β-thalassemia major, or HPFH - requires confirmation |
| AF | Prior blood transfusion - repeat at 4 months |
Summary: What Each Method Can and Cannot Do
| Feature | Alkaline Electrophoresis | Acid Electrophoresis | HPLC | Capillary Electrophoresis |
|---|---|---|---|---|
| Separates S from D/G/Lepore | No | Yes | No | Yes |
| Separates C from E | No (both at C) | Yes (E→A) | No (co-elute) | Yes |
| Quantifies HbA₂ accurately | No | No | Yes | Yes |
| Quantifies HbF accurately | No | No | Yes | Yes |
| Detects fast Hbs (H, Bart's) | Yes | - | Yes | Yes |
| Throughput/automation | Low | Low | High | High |
Clinical Approach to Microcytic Anemia with Hb Electrophoresis
CBC shows microcytic hypochromic anemia
↓
Check serum ferritin / iron studies
↓
┌─── Low ferritin ──→ Iron deficiency anemia (treat iron first)
│
└─── Normal/high ferritin ──→ Hb electrophoresis + HPLC
↓
┌── Elevated HbA₂ (≥4%) ──→ β-Thalassemia trait
│
├── Normal HbA₂, normal pattern ──→ α-Thalassemia trait (confirm by DNA)
│
├── HbS present ──→ Sickle syndromes (classify by pattern above)
│
├── HbC or HbE present ──→ See specific diagnosis above
│
└── Markedly elevated HbF, no/little HbA ──→ β-Thalassemia major
Sources: Tietz Textbook of Laboratory Medicine, 7th Edition, Ch. 77; Quick Compendium of Clinical Pathology, 5th Ed.; Harriet Lane Handbook, 23rd Ed.; Harrison's Principles of Internal Medicine 22E (2025)
This is a shared conversation. Sign in to Orris to start your own chat.