diabetes ketoacidosis
Reading File
Finding Sources
Searching PubMed
"diabetic ketoacidosis" management
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from two authoritative textbooks. Let me compile the full response.
Diabetic Ketoacidosis (DKA)
Definition
DKA is defined by three core biochemical features ("DKA triad"):
- Hyperglycemia - blood glucose elevated (or known history of diabetes)
- Ketonemia/ketonuria - urinary ketones ≥ 2+ or serum ketones ≥ 3.0 mmol/L
- Acidosis - arterial or venous pH < 7.3 (bicarbonate < 18 mmol/L)
- Goldman-Cecil Medicine, p. 2484
Pathophysiology
The core problem is insulin deficiency + counter-regulatory hormone excess (glucagon, cortisol, epinephrine, growth hormone).
Three interconnected mechanisms:
1. Hyperglycemia
- Insulin deficiency prevents glucose uptake into cells
- Glucagon stimulates hepatic gluconeogenesis and glycogenolysis
- Substrates flood the liver: amino acids + lactate from muscle, free fatty acids + glycerol from adipose tissue
- Glucose released into circulation faster than it can be utilized
2. Ketogenesis
- Insulin deficiency activates hormone-sensitive lipase in adipose tissue
- Free fatty acids (FFAs) surge in the bloodstream
- Liver converts long-chain FFAs to acetoacetate and beta-hydroxybutyrate (+ small amounts of acetone)
- These ketone bodies accumulate, causing high-anion-gap metabolic acidosis
3. Dehydration and electrolyte loss
- Hyperglycemia exceeds the renal threshold → osmotic diuresis
- Water, sodium, potassium, magnesium, calcium, and phosphorus are lost in urine
- Vomiting worsens fluid and electrolyte losses
- Fluid depletion → hemoconcentration → worsens hyperglycemia
Average deficits in severe DKA (per kg body weight):
| Deficit | Amount |
|---|---|
| Water | 70-120 mL/kg |
| Sodium | 8-10 mEq/kg |
| Potassium | 5-7 mEq/kg |
| Chloride | 6-8 mEq/kg |
| Phosphorus | ~3 mEq/kg |
- Rosen's Emergency Medicine, p. 2542
Important note on potassium: Despite total-body K+ depletion from osmotic diuresis, serum K+ may appear normal or even elevated at presentation due to acidosis (H+ shifts into cells, K+ shifts out). As insulin and fluids are given, K+ moves intracellularly and hypokalemia can become severe.
Precipitants
Most common:
- Infections (pneumonia, UTI, cellulitis)
- Inadequate insulin / non-adherence
- New-onset type 1 diabetes
- Acute coronary syndrome
- Unknown cause
Other precipitants:
-
Stroke, pulmonary embolism, acute pancreatitis
-
Alcohol intoxication
-
Endocrinopathies: Cushing syndrome, thyrotoxicosis, acromegaly
-
Drugs: corticosteroids, SGLT-2 inhibitors, clozapine, olanzapine, cocaine, lithium, sympathomimetics, thiazide diuretics
-
Severe burns, hyperthermia/hypothermia
-
Goldman-Cecil Medicine, Table 210-11
Clinical Features
Symptoms (develop over hours to days):
- Polyuria, polydipsia (classic hyperglycemia symptoms)
- Weakness, lethargy, nausea, anorexia
- Abdominal pain (can mimic acute abdomen)
- Vomiting
Signs:
- Dry skin and mucous membranes, decreased skin turgor
- Tachycardia, orthostatic hypotension
- Kussmaul breathing - deep, rapid respirations (compensatory respiratory alkalosis for metabolic acidosis)
- Fruity/acetone breath (from exhaled acetone)
- Depressed consciousness - ranges from lethargy to frank coma (correlates with hyperosmolality)
- Reduced jugular venous pressure
Diagnosis
| Parameter | Mild | Moderate | Severe |
|---|---|---|---|
| pH | 7.25-7.30 | 7.00-7.24 | < 7.00 |
| Bicarbonate | 15-18 mEq/L | 10-14 mEq/L | < 10 mEq/L |
| Anion gap | > 10 | > 12 | > 12 |
| Mental status | Alert | Drowsy | Stupor/Coma |
Key lab findings:
-
High anion gap metabolic acidosis - the anion gap rise is proportional to the bicarbonate fall
-
Pseudo-hyponatremia - measured serum Na+ is low due to osmotic shift of water from cells (correct: add 1.6 mEq/L Na+ for every 100 mg/dL glucose above normal)
-
Elevated WBC - can be due to metabolic acidosis itself, not necessarily infection
-
Elevated serum amylase - often non-pancreatic origin; can falsely suggest pancreatitis
-
Elevated hematocrit/Hgb - hemoconcentration
-
Prerenal azotemia (elevated BUN/Cr)
-
Goldman-Cecil Medicine, p. 2484
Management
Immediate priorities: Fluids, Insulin, Electrolytes
1. IV Fluids
- Adult fluid deficit is typically 3-5 L
- If in hypovolemic shock: give isotonic crystalloid (0.9% NaCl) as rapidly as possible (20 mL/kg boluses in children until SBP >80 mmHg)
- After stabilization: continue 0.9% NaCl or 0.45% NaCl at appropriate rate
- Switch to D5W / 0.45% NaCl when glucose drops to ≤ 300 mg/dL (prevents hypoglycemia while continuing insulin to clear ketones)
2. Insulin
- Regular insulin IV infusion: 0.1 units/kg/hour (standard protocol)
- Do not give insulin until potassium is ≥ 3.5 mEq/L (insulin drives K+ intracellularly and can cause fatal hypokalemia)
- Goal: glucose decrease of ~50-75 mg/dL/hour
- Recent evidence: subcutaneous insulin protocols are an alternative to IV infusion in mild-moderate DKA - a 2024 systematic review (PMID 39090718) found comparable outcomes between subcutaneous and IV insulin in adults; a 2026 meta-analysis (PMID 41208563) supports early addition of subcutaneous basal insulin during IV infusion to reduce rebound hyperglycemia
3. Potassium replacement
- If K+ < 3.5: replace K+ BEFORE starting insulin, 20-40 mEq/hr
- If K+ 3.5-5.0: add 20-30 mEq K+ to each liter of IV fluid
- If K+ > 5.0: hold potassium, monitor every 2 hours
4. Bicarbonate
- Generally NOT recommended unless pH < 6.9 (severe acidosis)
- Routine bicarbonate may worsen intracellular acidosis and hypokalemia
5. Phosphate
- Correct if severe depletion is present
6. Magnesium
- Correct with 1-2 g MgSO4 if depleted
Resolution criteria (transition to subcutaneous insulin):
- Blood glucose < 200 mg/dL
- Serum bicarbonate ≥ 15 mEq/L
- pH > 7.3
- Anion gap closed
Special Considerations
Intubation: Avoid if possible - DKA patients have a tremendous respiratory drive; matching ventilator settings to this is difficult. If intubation is required (comatose/vomiting patient), maintain hyperventilation to prevent worsening acidosis.
SGLT-2 inhibitors: Can cause euglycemic DKA - DKA with near-normal blood glucose levels. Important not to miss.
DKA vs. HHS (Hyperosmolar Hyperglycemic State): HHS has more severe hyperglycemia but less ketosis/acidosis, because residual insulin (in type 2 DM) suppresses lipolysis. Some patients present with a mixed picture.
Monitoring: Maintain meticulous flow sheets tracking - vital signs, fluid I/O, serum glucose, K+, Cl-, HCO3-, pH, and insulin given.
Sources: Goldman-Cecil Medicine (International Edition) | Rosen's Emergency Medicine | Recent PubMed evidence (PMIDs 39090718, 41208563)
ketogenisis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Excellent - both diagrams retrieved. Here is the comprehensive answer:
Ketogenesis
Overview
Ketogenesis is the metabolic process by which the liver produces ketone bodies from fatty acids. It occurs primarily in hepatic mitochondria under conditions of high fatty acid oxidation - starvation, prolonged fasting, low-carbohydrate states, or insulin deficiency (as in DKA).
The three ketone bodies produced are:
- Acetoacetate (primary product)
- Beta-hydroxybutyrate (3-hydroxybutyrate) (most abundant in blood)
- Acetone (minor, volatile - exhaled via lungs)
Normal blood ketone concentration: < 0.2 mmol/L. The liver produces ketones but does not use them; extrahepatic tissues (brain, heart, muscle, kidney) consume them as fuel.
- Harper's Illustrated Biochemistry, 32nd Ed, p. 232
Why Does Ketogenesis Happen? (The Setup)
During fasting or insulin deficiency, the liver is flooded with free fatty acids (FFAs) mobilized from adipose tissue. This creates a biochemical dilemma:
- Acetyl-CoA overload from beta-oxidation of fatty acids accumulates faster than the TCA cycle can handle
- OAA (oxaloacetate) is depleted - because:
- Fatty acid oxidation raises NADH, which shifts OAA to malate
- OAA is consumed by gluconeogenesis (liver prioritizes making glucose)
- Without OAA, acetyl-CoA cannot enter the TCA cycle (citrate synthase reaction is blocked)
- The excess acetyl-CoA is therefore shunted into ketogenesis
- Lippincott's Illustrated Reviews: Biochemistry, 8th Ed, p. 555
The Ketogenesis Pathway (Step by Step)
All reactions occur in hepatic mitochondria.
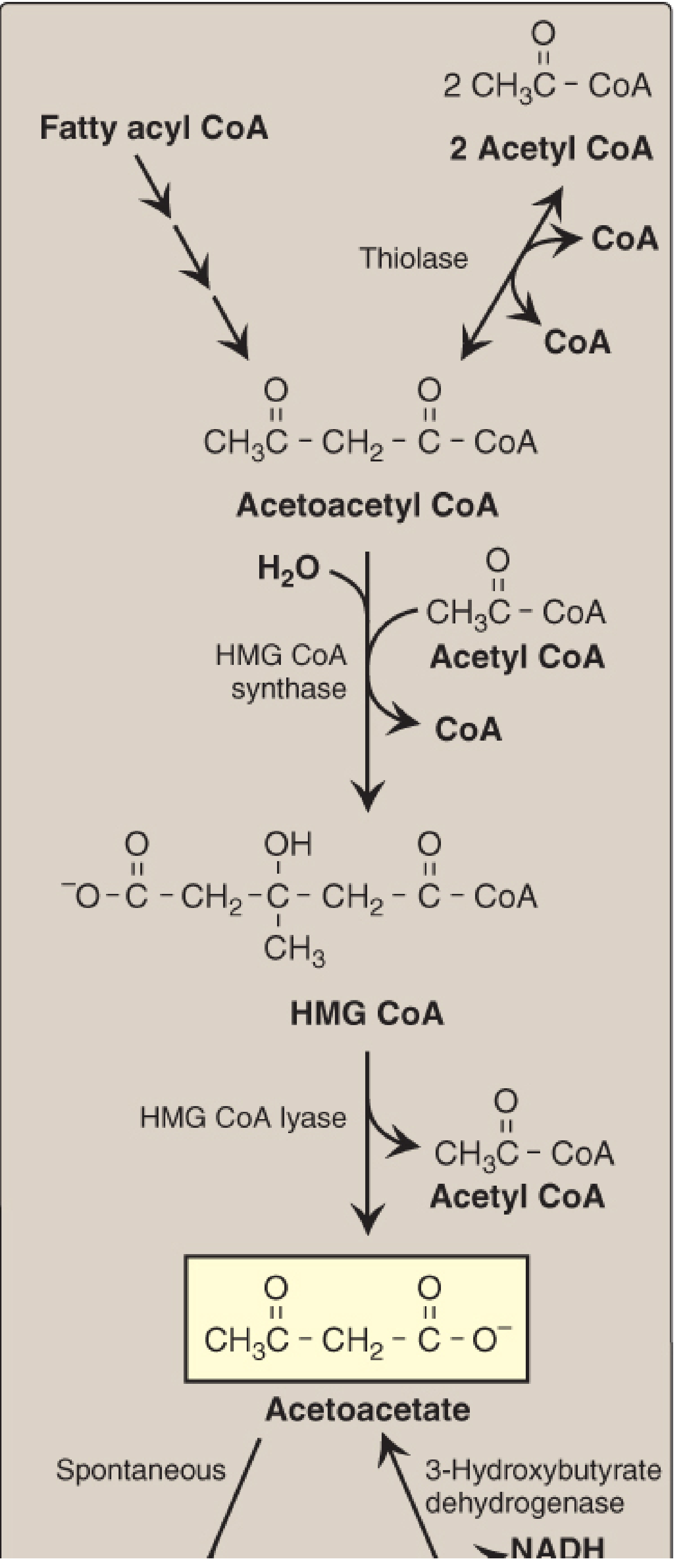
Step 1: Acetoacetyl-CoA formation
- 2 molecules of acetyl-CoA condense
- Enzyme: Thiolase (reversal of the last step of beta-oxidation)
- Product: Acetoacetyl-CoA
Step 2: HMG-CoA formation (rate-limiting step)
- Acetoacetyl-CoA + another acetyl-CoA + H₂O
- Enzyme: Mitochondrial HMG-CoA synthase (rate-limiting, liver-specific)
- Product: 3-Hydroxy-3-methylglutaryl CoA (HMG-CoA)
- Note: HMG-CoA synthase is found in significant quantities only in the liver - this is why only the liver produces ketone bodies in quantity
Step 3: Acetoacetate formation
- HMG-CoA is cleaved
- Enzyme: HMG-CoA lyase
- Products: Acetoacetate + acetyl-CoA
- The released CoA is important - it is recycled back to support continued fatty acid oxidation
Step 4a: Beta-hydroxybutyrate formation
- Acetoacetate + NADH + H⁺ → 3-hydroxybutyrate + NAD⁺
- Enzyme: 3-Hydroxybutyrate dehydrogenase (mitochondrial)
- The equilibrium is driven toward 3-hydroxybutyrate when the NAD⁺/NADH ratio is low (i.e., during active fatty acid oxidation), so 3-HB is the predominant ketone in DKA
Step 4b: Acetone formation
- Acetoacetate spontaneously decarboxylates → acetone + CO₂
- Not enzymatic; acetone is metabolically inert and exhaled (fruity breath in DKA)
Three Key Regulatory Steps
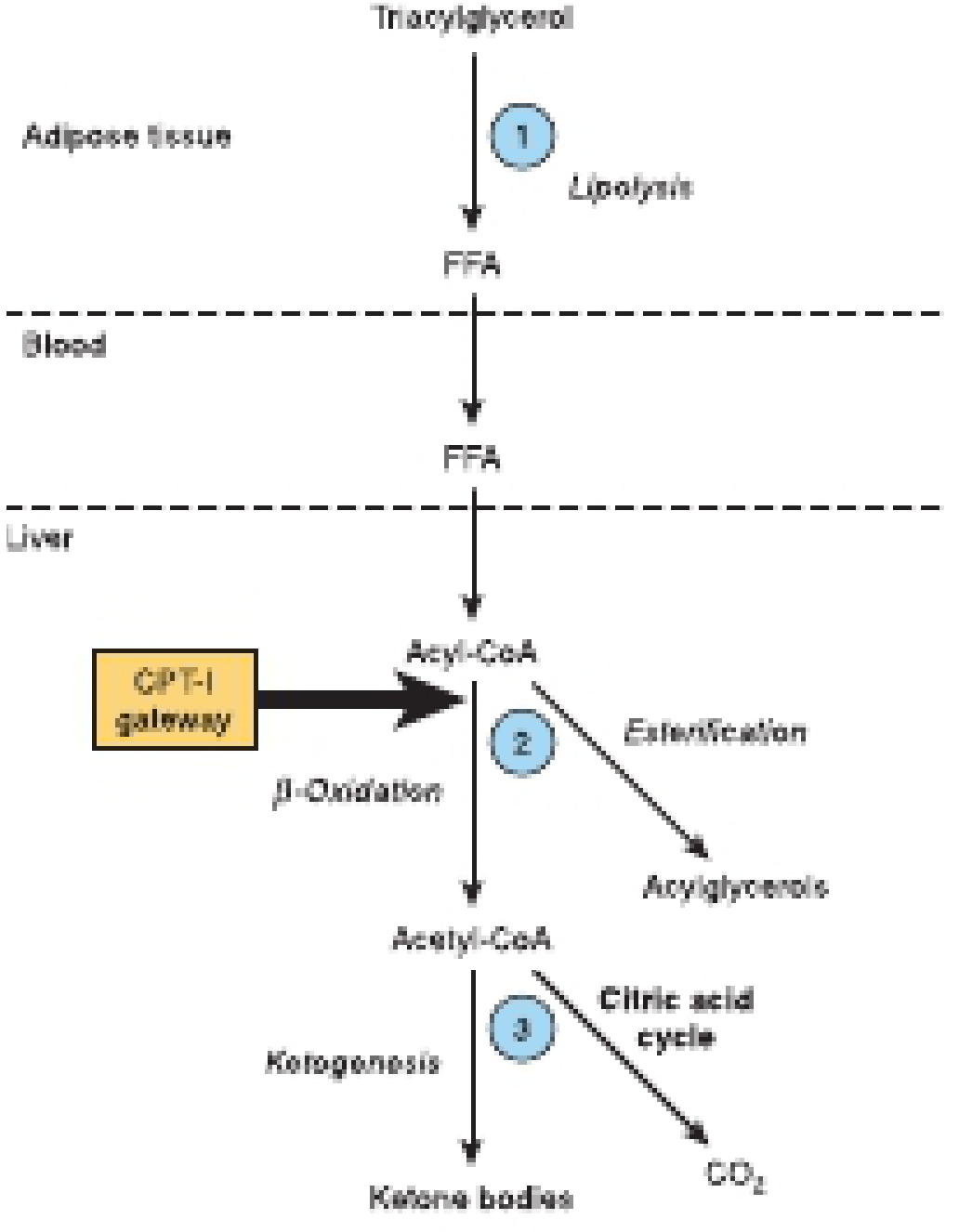
Step 1 - Lipolysis in adipose tissue:
- Ketosis cannot occur unless FFAs are mobilized from adipose triacylglycerol
- Insulin inhibits hormone-sensitive lipase → suppresses lipolysis
- Glucagon/epinephrine activate lipolysis
- Low insulin (fasting, DKA) = FFA flood → ketogenesis substrate available
Step 2 - CPT-I gateway into mitochondria:
- Carnitine palmitoyl transferase I (CPT-I) transports long-chain acyl-CoA into mitochondria for beta-oxidation
- Malonyl-CoA (the first intermediate in fatty acid synthesis) is a potent inhibitor of CPT-I
- In the fed state: high insulin → high malonyl-CoA → CPT-I inhibited → fatty acids go to esterification, not oxidation
- In fasting/DKA: low insulin → malonyl-CoA falls → CPT-I active → beta-oxidation and ketogenesis increase
Step 3 - Acetyl-CoA: TCA cycle vs. ketogenesis:
-
As FFA flux increases, acetyl-CoA outpaces the TCA cycle capacity (OAA is depleted)
-
Proportionately more acetyl-CoA is diverted into ketogenesis
-
At very high FFA concentrations, essentially all excess acetyl-CoA goes to ketone bodies
-
Harper's Illustrated Biochemistry, 32nd Ed, p. 234-235
Ketolysis (Use of Ketones by Peripheral Tissues)
Peripheral tissues (brain, heart, skeletal muscle, kidney) use ketones as energy fuel. The liver cannot use its own ketone bodies because it lacks succinyl-CoA transferase (thiophorase) - the enzyme needed for the first step of ketolysis.
Ketolysis steps:
- 3-Hydroxybutyrate → Acetoacetate (by 3-HB dehydrogenase)
- Acetoacetate + succinyl-CoA → Acetoacetyl-CoA + succinate (by succinyl-CoA transferase - absent in liver)
- Acetoacetyl-CoA → 2 Acetyl-CoA (by thiolase)
- Acetyl-CoA enters TCA cycle → ATP production
During prolonged starvation, the brain adapts to use ketones as its primary fuel, reducing its dependence on glucose.
- Lippincott's Illustrated Reviews: Biochemistry, 8th Ed, p. 556
Physiological vs. Pathological Ketogenesis
| State | FFA Level | Insulin | Ketone Level | Significance |
|---|---|---|---|---|
| Well-fed | Low | High | < 0.2 mmol/L | Negligible |
| Fasting (overnight) | Moderate | Low-normal | 0.2-0.5 mmol/L | Normal |
| Prolonged starvation | High | Very low | 1-7 mmol/L | Physiological ketosis |
| DKA | Very high | Absent | > 3-5 mmol/L | Pathological - acidosis |
| Ketogenic diet | Moderate-high | Low | 0.5-3 mmol/L | Deliberate nutritional ketosis |
In DKA, the process is extreme: no insulin means maximal lipolysis, maximal CPT-I activity, minimal TCA cycle flux (OAA depleted for gluconeogenesis), and maximal ketone production. The buffering capacity of blood is overwhelmed, leading to acidosis (pH < 7.3).
Key Enzymes Summary
| Enzyme | Location | Role | Rate-Limiting? |
|---|---|---|---|
| Hormone-sensitive lipase | Adipose | Liberates FFA from triglycerides | Yes (step 1 control) |
| CPT-I | Outer mitochondrial membrane | Transports acyl-CoA into mitochondria | Yes (step 2 control) |
| Thiolase | Mitochondria | Condenses 2 acetyl-CoA → acetoacetyl-CoA | No |
| Mitochondrial HMG-CoA synthase | Mitochondria | Acetoacetyl-CoA + acetyl-CoA → HMG-CoA | Yes (committed step) |
| HMG-CoA lyase | Mitochondria | HMG-CoA → acetoacetate + acetyl-CoA | No |
| 3-Hydroxybutyrate dehydrogenase | Mitochondria | Acetoacetate ↔ 3-hydroxybutyrate | No (equilibrium enzyme) |
Sources: Harper's Illustrated Biochemistry (32nd Ed) | Lippincott's Illustrated Reviews: Biochemistry (8th Ed)
vit A
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I have comprehensive content from three authoritative sources. Here is the full answer:
Vitamin A
Forms and Chemical Structure
Vitamin A is a fat-soluble vitamin. The term covers a group of related compounds:
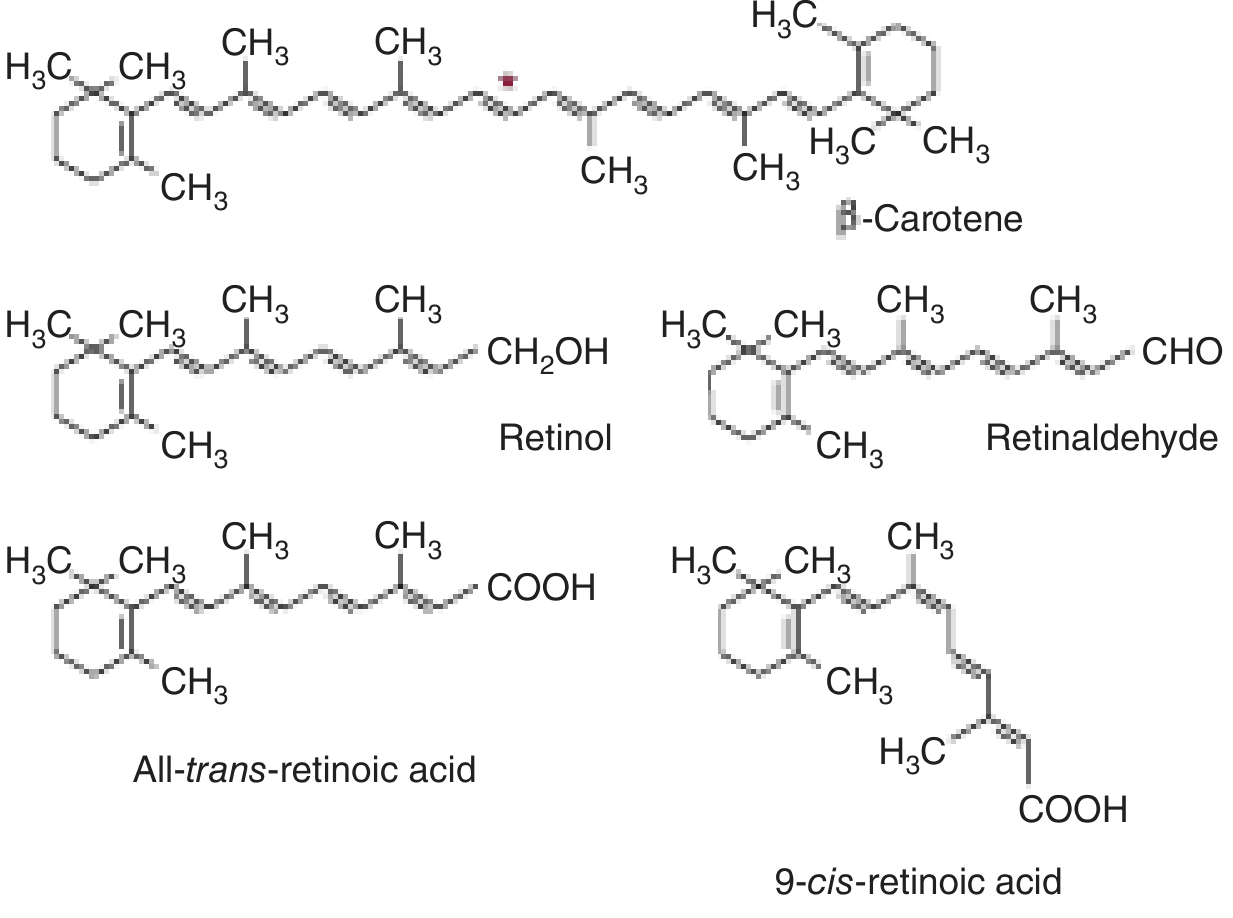
| Form | Chemical group | Role |
|---|---|---|
| Retinol | Alcohol (-CH₂OH) | Transport and storage form |
| Retinal (retinaldehyde) | Aldehyde (-CHO) | Visual cycle |
| Retinoic acid | Carboxylic acid (-COOH) | Gene regulation |
| Beta-carotene | Provitamin (plant source) | Converted to retinol in gut |
The term retinoids covers all natural and synthetic compounds structurally related to vitamin A.
- Harper's Illustrated Biochemistry, 32nd Ed
Dietary Sources and Units
Animal sources (preformed vitamin A - retinyl esters):
- Liver (richest source), fish liver oils, eggs, milk, butter
Plant sources (provitamin A carotenoids):
- Yellow/orange vegetables: carrots, squash, sweet potato
- Dark leafy greens: spinach, kale
- The most important is beta-carotene, symmetrically cleaved by carotene dioxygenase to yield 2 molecules of retinaldehyde
Measurement - Retinol Activity Equivalents (RAE):
-
1 RAE = 1 µg retinol = 12 µg β-carotene = 24 µg other provitamin A carotenoids
-
Old units: 1 IU = 0.3 µg retinol (still seen on food labels)
-
Carotenoids contribute ~30% of dietary vitamin A in humans
-
Harrison's Internal Medicine, 22nd Ed, p. 2654
Absorption and Metabolism
-
Digestion: Retinyl esters (animal sources) are hydrolyzed to retinol in the intestinal lumen. Beta-carotene is cleaved to retinaldehyde then reduced to retinol in intestinal mucosal cells. Absorption requires bile salts, pancreatic enzymes, and antioxidants (fat-soluble vitamin)
-
Transport to liver: Retinol is packaged into chylomicrons and transported via lymph to the circulation, then taken up by the liver via apolipoprotein E receptors
-
Liver storage: >90% of body's vitamin A is stored in the liver - specifically in perisinusoidal stellate (Ito) cells as retinyl esters. Reserves last at least 6 months on adequate diet
-
Mobilization: For release from the liver, retinol binds to retinol-binding protein (RBP), which then associates with transthyretin (prealbumin) to form a trimolecular complex. This:
- Prevents glomerular filtration of retinol (protects against toxicity)
- Allows recognition by cell-surface RBP receptors on peripheral tissues
-
Cellular uptake: After internalization, retinol binds cellular retinol-binding proteins (CRBPs) for transport to nucleus. Can be stored as retinyl ester or oxidized to retinoic acid
Note: Zinc deficiency impairs mobilization of vitamin A from liver stores. Alcohol interferes with vitamin A metabolism.
- Robbins & Kumar Basic Pathology, p. 292 | Harrison's 22nd Ed
Functions
1. Vision (Retinal)
The visual cycle depends entirely on vitamin A in the form of retinaldehyde:
- In the pigment epithelium: all-trans-retinol → isomerized to 11-cis-retinol → oxidized to 11-cis-retinaldehyde
- 11-cis-retinaldehyde binds to opsin (via a Schiff base to a lysine residue) to form rhodopsin (in rods) or iodopsins (in cones - 3 types for color)
- On light absorption: 11-cis-retinaldehyde isomerizes to all-trans-retinaldehyde → conformational change in opsin → triggers a guanine nucleotide amplification cascade → nerve impulse
- Rhodopsin is hydrolyzed; all-trans-retinaldehyde is released and recycled
In deficiency: Both dark adaptation time and the ability to see in dim light are impaired - this is night blindness (nyctalopia), the earliest symptom.
- Harper's Illustrated Biochemistry, 32nd Ed
2. Epithelial Cell Differentiation (Retinoic Acid)
Retinoic acid is a nuclear hormone receptor ligand that acts like a steroid hormone:
- Binds Retinoic Acid Receptors (RARs) and Retinoid X Receptors (RXRs)
- RAR/RXR heterodimers bind Retinoic Acid Response Elements (RAREs) in target gene promoters
- Regulates: epithelial differentiation, cell-cycle control, growth factor receptor expression, tumor suppressor genes
Key role: maintaining mucus-secreting columnar epithelium. Without vitamin A, epithelial cells undergo squamous metaplasia - differentiating into keratinizing epithelium instead.
RXR also heterodimerizes with:
- Vitamin D receptor (VDR)
- Thyroid hormone receptor (TR)
- PPARs (peroxisome proliferator-activated receptors) - regulates fatty acid and carbohydrate metabolism; basis for thiazolidinedione drugs (rosiglitazone, pioglitazone)
3. Immune Function
Vitamin A supplementation reduces mortality in deficient children by 20-30% by improving immune function, particularly resistance to infections.
4. Lipid Metabolism
Retinoids inhibit adipogenesis and stimulate lipid breakdown. RXR-PPAR interactions regulate fat metabolism.
- Robbins & Kumar Basic Pathology, p. 293 | Harrison's 22nd Ed
Deficiency
Causes
- Dietary (most common worldwide): inadequate intake of animal products and vegetables, especially in children in developing countries
- Malabsorption: celiac disease, Crohn disease, colitis, bariatric surgery, prolonged mineral oil laxative use
- Concurrent zinc deficiency: impairs hepatic mobilization of vitamin A
Clinical Manifestations (in order of appearance)
Eyes (most important):
- Night blindness - earliest symptom; impaired rod function in dim light
- Xerosis conjunctivae - dryness of the conjunctiva; normal lacrimal/mucus epithelium replaced by keratinized epithelium
- Bitot spots - small opaque plaques of keratin debris on conjunctiva (pathognomonic)
- Keratomalacia - softening and destruction of the cornea → blindness
The full eye picture is called xerophthalmia (dry eye).
Other organs:
-
Respiratory tract: squamous metaplasia of bronchial epithelium → loss of mucociliary clearance → recurrent pulmonary infections
-
Urinary tract: keratin debris desquamation → renal/bladder stones
-
Skin: follicular/papular dermatosis (hyperkeratosis of hair follicles - "phrynoderma")
-
Immune system: increased susceptibility to infections (measles, diarrheal illness)
-
Robbins & Kumar Basic Pathology, p. 293
Toxicity (Hypervitaminosis A)
Since vitamin A is fat-soluble, it accumulates and can reach toxic levels. Unlike beta-carotene (excess just causes orange skin - carotenodermia, harmless), preformed vitamin A is toxic.
Acute Toxicity
- Headache, dizziness, vomiting, stupor, blurred vision
- Can mimic pseudotumor cerebri (raised intracranial pressure)
- Historical note: Arctic explorers developed acute toxicity from eating polar bear liver (extremely rich in vitamin A)
Chronic Toxicity
- Weight loss, anorexia, nausea, vomiting
- Bone and joint pain
- Retinoic acid stimulates osteoclast activity → increased bone resorption → high fracture risk
- Hepatotoxicity (liver fibrosis with very high chronic intake)
Teratogenicity
-
Retinoids are potent teratogens - cause craniofacial, cardiovascular, thymic, and CNS malformations
-
Isotretinoin (synthetic retinoid for acne) is strictly contraindicated in pregnancy
-
Vitamin A supplements >10,000 IU/day should be avoided in pregnancy
-
Robbins & Kumar Basic Pathology, p. 293
Summary Table
| Feature | Detail |
|---|---|
| Type | Fat-soluble vitamin |
| Active forms | Retinol, retinal, retinoic acid |
| Provitamin | Beta-carotene (plant) |
| Storage | Liver stellate cells (>90%) |
| Transport protein | Retinol-binding protein (RBP) + transthyretin |
| Vision role | 11-cis-retinal in rhodopsin/iodopsins |
| Gene regulation | RAR/RXR nuclear receptors |
| RDA | ~700-900 µg RAE/day (adult) |
| Deficiency sign | Night blindness → xerophthalmia → blindness |
| Toxicity | Pseudotumor cerebri, bone resorption, teratogenicity |
Sources: Robbins & Kumar Basic Pathology | Harrison's Principles of Internal Medicine (22nd Ed) | Harper's Illustrated Biochemistry (32nd Ed)
vit A
The previous response covered Vitamin A comprehensively - here's a quick recap of the key points:
Vitamin A - Quick Reference
Forms
| Form | Function |
|---|---|
| Retinol | Transport/storage |
| Retinal | Vision (rhodopsin) |
| Retinoic acid | Gene regulation via RAR/RXR |
| Beta-carotene | Provitamin (plant source) |
Key Functions
- Vision - 11-cis-retinal forms rhodopsin in rods; iodopsins in cones
- Epithelial differentiation - maintains mucus-secreting epithelium via nuclear RAR/RXR receptors
- Immune function - supplementation reduces child mortality 20-30% in deficient populations
- Lipid metabolism - RXR dimerizes with PPARs
Deficiency (in order)
- Night blindness → Xerosis conjunctivae → Bitot spots → Keratomalacia → Blindness
- Squamous metaplasia of respiratory/urinary epithelium
- Follicular dermatosis (phrynoderma)
Toxicity
- Acute: Headache, pseudotumor cerebri, vomiting
- Chronic: Bone resorption, fractures, hepatotoxicity
- Pregnancy: Potent teratogen - isotretinoin strictly contraindicated
Storage & Transport
-
90% stored in hepatic stellate (Ito) cells
- Transported bound to RBP + transthyretin
Is there a specific aspect of Vitamin A you'd like me to go deeper on - such as the visual cycle mechanism, retinoic acid receptor signaling, clinical management of deficiency, or use of retinoids in dermatology/oncology?
This is a shared conversation. Sign in to Orris to start your own chat.