Here are all 9 comprehensive 10-marks answers:
A) MALIGNANT MELANOMA
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Malignant melanoma is a malignant neoplasm arising from melanocytes, most commonly at the dermo-epidermal junction (DEJ). It is the most lethal of all skin cancers, with a strong tendency for early metastasis.
EPIDEMIOLOGY
- Incidence rising in light-skinned populations worldwide
- Peak incidence: 4th–5th decade
- Rare in dark-skinned races; acral and mucosal types more common in Asians and Africans
- Children rarely affected; poor prognosis when they are
RISK FACTORS
| Category | Factors |
|---|
| Phenotypic | Light complexion, light eyes, blond/red hair, poor tanning, heavy freckling |
| UV | Blistering sunburns in childhood, PUVA, tanning lamps |
| Nevus-related | >50 common nevi, dysplastic nevi, giant congenital naevus |
| Genetic | Family history of melanoma, BRAF mutation (non-sun-exposed skin), KIT mutation (acral/mucosal), CDKN2A/p16 mutation |
| Systemic | Immunodeficiency, xeroderma pigmentosum, burn scars |
CLINICAL TYPES (Clinicopathologic Classification)
| Type | Features |
|---|
| Superficial Spreading Melanoma (SSM) | Most common (70%); flat, asymmetric, variegated plaque; long radial growth phase; arises in pre-existing nevi |
| Nodular Melanoma (NM) | Second most common (15–30%); rapid vertical growth; blue-black nodule; worst prognosis at diagnosis |
| Lentigo Maligna Melanoma (LMM) | Sun-damaged skin of elderly (face/neck); long in-situ phase (lentigo maligna → invasion = LMM) |
| Acral Lentiginous Melanoma (ALM) | Palms, soles, subungual; most common type in dark-skinned individuals; associated with KIT mutations |
| Desmoplastic Melanoma | Head/neck; amelanotic; deeply invasive; high neurotropism; high local recurrence |
| Mucosal Melanoma | Oral, nasal, genital mucosae; poor prognosis |
| Amelanotic Melanoma | Lacks pigment; pink papule; diagnosis often delayed |
ABCDE CRITERIA (Screening Tool)
| Letter | Criterion |
|---|
| A | Asymmetry |
| B | Border irregularity |
| C | Colour variegation (multiple shades) |
| D | Diameter >6 mm |
| E | Evolution (change in size, shape, colour) |
"Ugly Duckling" sign — any mole looking different from others in the same patient
HISTOPATHOLOGY
Radial growth phase (RGP): Melanocytes proliferate horizontally at DEJ and epidermis; no metastatic potential.
Vertical growth phase (VGP): Tumour nodule invades dermis; metastatic potential begins.
Histological features:
- Asymmetry and lack of maturation with depth
- Buckshot (pagetoid) scatter of melanocytes in epidermis
- Irregular junctional nests at rete ridges and suprapapillary plates
- Absent "maturation" (nests fail to become smaller in deeper dermis)
- Dermal mitoses, lymphoid infiltrate, satellite metastases
STAGING
Clark's Level (depth of invasion):
| Level | Invasion |
|---|
| I | Intraepidermal (in situ) |
| II | Into papillary dermis |
| III | Filling papillary dermis |
| IV | Into reticular dermis |
| V | Into subcutaneous fat |
Breslow Thickness (most important prognostic factor):
- ≤1.0 mm → thin (low risk)
- 1.01–2.0 mm → intermediate
- 2.01–4.0 mm → thick
-
4.0 mm → very thick (high risk)
AJCC TNM Staging:
- T = tumour thickness + ulceration + mitotic rate
- N = nodal involvement (SLN biopsy)
- M = distant metastases (M1c = visceral; worst prognosis)
INVESTIGATIONS
- Dermoscopy — most useful non-invasive tool
- Excision biopsy — complete removal with 1–3 mm margin (saucerisation technique)
- Sentinel lymph node biopsy (SLNB) — for tumours >1 mm or with ulceration
- Imaging: PET-CT, CT chest/abdomen/pelvis for staging
- Mutation testing: BRAF V600E, KIT, NRAS (guides targeted therapy)
- LDH — elevated in metastatic disease (poor prognostic marker)
TREATMENT
Surgery (Primary Treatment):
| Breslow Thickness | Excision Margin |
|---|
| In situ | 5 mm |
| ≤1.0 mm | 1 cm |
| 1.01–2.0 mm | 1–2 cm |
| >2.0 mm | 2 cm |
Adjuvant Therapy (Stage III–IV):
| Drug | Target/Class |
|---|
| Dabrafenib + Trametinib | BRAF V600E + MEK inhibitors |
| Vemurafenib | BRAF V600E inhibitor |
| Pembrolizumab / Nivolumab | Anti-PD1 (checkpoint inhibitors) |
| Ipilimumab | Anti-CTLA4 |
| Interferon-α | Adjuvant (older regimens) |
Radiation: For lentigo maligna (inoperable), brain metastases, palliative
Isolated Limb Perfusion: Melphalan for unresectable in-transit metastases
PROGNOSIS
- 5-year survival: Stage I = 97%; Stage II = 68%; Stage III = 63%; Stage IV = 23%
- Ulceration, high mitotic rate, nodal involvement = poor prognostic factors
- Pregnancy does not worsen survival but increases recurrence
Source: Andrews' Diseases of the Skin; Fitzpatrick's Dermatology 9e
C) DERMATOLOGICAL EMERGENCIES
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Dermatological emergencies are life-threatening conditions primarily or secondarily involving the skin, requiring immediate diagnosis and management to prevent death or serious morbidity.
CLASSIFICATION
I. Severe Cutaneous Adverse Drug Reactions (SCARs)
- Stevens-Johnson Syndrome (SJS)
- Toxic Epidermal Necrolysis (TEN)
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS/DiHS)
- Acute Generalised Exanthematous Pustulosis (AGEP)
II. Blistering/Exfoliative Emergencies
- Staphylococcal Scalded Skin Syndrome (SSSS)
- Pemphigus vulgaris (severe/crisis)
- Erythroderma / Exfoliative Dermatitis
III. Vascular/Infectious Emergencies
- Necrotising Fasciitis
- Meningococcaemia / Purpura Fulminans
- Toxic Shock Syndrome (TSS)
IV. Miscellaneous
- Angioedema / Anaphylaxis
- Calciphylaxis
KEY CONDITIONS IN DETAIL
1. Stevens-Johnson Syndrome / Toxic Epidermal Necrolysis
Definition: SCARs characterised by epidermal detachment and necrosis.
- SJS: <10% body surface area (BSA) detachment
- SJS-TEN overlap: 10–30% BSA
- TEN: >30% BSA detachment
Causes: Drugs — sulfonamides, anticonvulsants (carbamazepine, phenytoin, lamotrigine), allopurinol, NSAIDs, antibiotics; infections (Mycoplasma → SJS in children)
Pathogenesis:
- Drug-specific CD8⁺ T cell activation → keratinocyte apoptosis via FasL, perforin/granzyme B, TNF-α, and granulysin (principal mediator)
- HLA associations: HLA-B5701 (abacavir), HLA-B1502 (carbamazepine in Asians)
Clinical Features:
- Prodrome: fever, malaise, photophobia, painful skin
- Skin: erythema → bullae → sheet-like epidermal detachment; Nikolsky sign positive
- Mucosal involvement: oral, ocular, genital mucosae (distinguishes from SSSS)
- Target-like lesions; atypical targets (flat, 2 zones, no central blister)
- Positive "Asboe-Hansen" sign (lateral pressure extends bulla)
SCORTEN (Severity Score):
| Parameter | Score |
|---|
| Age >40 | 1 |
| Heart rate >120 | 1 |
| BSA detachment >10% | 1 |
| BUN >28 mg/dL | 1 |
| Glucose >252 mg/dL | 1 |
| Bicarbonate <20 mEq/L | 1 |
| Malignancy | 1 |
Score 0–1 = 3.2% mortality → Score ≥5 = >90% mortality
Management:
- Stop offending drug immediately
- Admit to ICU/burns unit
- Fluid and electrolyte replacement (Parkland-like formula)
- Nutritional support (NG tube)
- Wound care: non-adherent dressings (petroleum-impregnated gauze)
- Ophthalmology consult (eye drops, lysis of synechiae)
- Specific treatments:
- IVIG (0.5–1 g/kg/day × 3 days) — most used
- Cyclosporine (3–5 mg/kg/day) — recent evidence supports
- Systemic corticosteroids — controversial; avoid in established TEN
- Etanercept (25–50 mg) — emerging evidence
- Avoid NSAIDs (antipyretics: paracetamol only)
- Treat secondary infections (no prophylactic antibiotics)
2. DRESS Syndrome (Drug Reaction with Eosinophilia and Systemic Symptoms)
Also called: Drug-Induced Hypersensitivity Syndrome (DiHS)
Causative drugs: Anticonvulsants (phenytoin, carbamazepine, phenobarbital), allopurinol, dapsone, minocycline, sulfonamides
Pathogenesis: Defective detoxification of reactive drug metabolites → T cell activation; reactivation of HHV-6, HHV-7, CMV, EBV (sequential viral reactivation)
Clinical features:
- Onset: 2–8 weeks after drug initiation (long latency — key distinguisher)
- Morbilliform/erythrodermic rash + facial oedema (characteristic)
- Eosinophilia, atypical lymphocytosis
- Systemic: hepatitis (most common), pneumonitis, myocarditis, nephritis, thyroiditis
- RegiSCAR score used for diagnosis
Management: Stop drug; systemic corticosteroids (0.5–1 mg/kg prednisolone); taper slowly over 3–6 months (rapid taper → rebound); IVIG for refractory cases; monitor LFTs, renal function, TSH
3. Erythroderma / Exfoliative Dermatitis
Definition: Generalised erythema and scaling involving >90% BSA.
Causes (PSMED mnemonic):
- Psoriasis (most common in adults)
- Seborrhoeic dermatitis
- Malignancy (Sézary syndrome, lymphoma)
- Eczema/atopic dermatitis
- Drugs
Complications: Hypothermia, fluid loss, hypoalbuminaemia, high-output cardiac failure, infection, DVT
Management: Admit; temperature regulation; fluid resuscitation; emollients; treat underlying cause; systemic steroids (non-psoriasis); methotrexate/cyclosporine (psoriatic)
4. Necrotising Fasciitis
Definition: Rapidly progressive, life-threatening soft tissue infection along fascial planes with extensive necrosis.
Types:
- Type I (polymicrobial): Fournier's gangrene (perineum/genitalia)
- Type II (Group A Streptococcus): "flesh-eating bacteria"
Features: Disproportionate pain, swelling, "woody" hard skin, crepitus, systemic toxicity, skin necrosis; gas on imaging
Management: Urgent surgical debridement (repeated as needed); broad-spectrum antibiotics (pip-tazo + clindamycin + vancomycin); IVIG; hyperbaric oxygen (adjunct)
LRINEC Score (Laboratory Risk Indicator for Necrotising Fasciitis): CRP, WBC, Hb, Na⁺, creatinine, glucose — score ≥6 = high risk
5. Meningococcaemia / Purpura Fulminans
- Non-blanching petechiae/purpura ± haemorrhagic bullae in context of fever/sepsis
- Disseminated Intravascular Coagulation (DIC) → purpura fulminans
- Management: Immediate parenteral benzylpenicillin/ceftriaxone before hospital transfer; ICU; FFP; heparin controversial
KEY EXAM TABLE — Drug Reactions Comparison
| Feature | SJS/TEN | DRESS | AGEP |
|---|
| Latency | 1–3 weeks | 2–8 weeks | 1–5 days |
| Mucosal involvement | Yes (prominent) | Yes (mild) | Rare |
| Pustules | No | No | Yes (sterile) |
| Eosinophilia | Mild | Marked | Variable |
| Causative drugs | Anticonvulsants, sulfa | Same + allopurinol | Antibiotics, CCBs |
| Mortality | 10–30% (TEN) | 5–10% | <5% |
Sources: Fitzpatrick's Dermatology 9e; Andrews' Diseases of the Skin
D) KAPOSI SARCOMA
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Kaposi sarcoma (KS) is a vascular and lymphatic neoplasm caused by Human Herpesvirus-8 (HHV-8), also called Kaposi Sarcoma-associated Herpesvirus (KSHV). It is characterised by multifocal, violaceous skin lesions and can involve viscera.
AETIOLOGY / PATHOGENESIS
Causative agent: HHV-8 (KSHV) — a gamma-herpesvirus
Pathogenesis:
- HHV-8 infects endothelial cells and B lymphocytes
- Viral oncoproteins (v-cyclin, v-FLIP, LANA) subvert cell cycle control, inhibit apoptosis
- VEGF and inflammatory cytokines (IL-6, bFGF, oncostatin M) drive angiogenesis and spindle cell proliferation
- Immunosuppression (HIV, iatrogenic) allows unrestricted HHV-8 replication
- HHV-8 also activates NF-κB pathway → anti-apoptotic signalling
TYPES OF KAPOSI SARCOMA
| Type | Population | HHV-8 | Clinical Behaviour |
|---|
| Classic / Mediterranean | Elderly men of Eastern European/Mediterranean origin | Yes | Indolent; lower extremity lesions; rarely systemic |
| Endemic / African | Sub-Saharan Africa; two forms: adult (nodular) and children (lymphadenopathic) | Yes | Aggressive in children (lymphadenopathy > skin) |
| Iatrogenic / Transplant-associated | Organ transplant recipients on immunosuppression | Yes | Resolves/improves on reducing immunosuppression |
| AIDS-related / Epidemic | HIV-positive individuals (especially MSM) | Yes | AIDS-defining illness; widespread; visceral involvement common |
CLINICAL FEATURES
Skin lesions progress: Patch → Plaque → Nodule/Tumour
| Stage | Appearance |
|---|
| Patch | Pink-red to violaceous flat macule; subtle telangiectasias |
| Plaque | Raised, violaceous, firm; poorly defined |
| Nodule/Tumour | Purple-black, firm nodule; may ulcerate; lymphoedema |
Distribution:
- AIDS-KS: Face (nose, periorbital), oral mucosa (hard palate — pathognomonic), extremities, trunk; follows Langer's lines
- Classic KS: Lower extremities bilaterally
- Any organ may be involved: lungs (haemoptysis), GI tract (bleeding), lymph nodes
Classic KS: Violaceous/brownish plaques and nodules on the lower extremities of elderly men; chronic, indolent course; lymphoedema common.
Oral KS (hard palate): Almost pathognomonic of AIDS-related KS; red to violaceous flat or raised lesion.
AIDS CLINICAL TRIALS GROUP (ACTG) STAGING
| Parameter | Good Risk (T0, I0, S0) | Poor Risk (T1, I1, S1) |
|---|
| T (Tumour) | Skin ± lymph nodes, no oral KS | Tumour-associated oedema/ulceration, extensive oral, visceral |
| I (Immune) | CD4 ≥150/μL | CD4 <150/μL |
| S (Systemic) | No systemic illness, no B symptoms | B symptoms (fever, weight loss >10%), other OIs |
HISTOPATHOLOGY
Three stages on biopsy:
- Patch stage: Irregular vascular spaces lined by bland endothelium; plasma cells and RBC extravasation
- Plaque stage: Spindle cells between vascular spaces; haemosiderin deposits; hyaline globules (PAS+)
- Nodular stage: Dense fascicles of spindle cells (tumour cells); slit-like vascular spaces; mitoses; haemosiderin
IHC: HHV-8 LANA (Latency-Associated Nuclear Antigen) staining — gold standard; CD31, CD34 (endothelial markers) positive
DIAGNOSIS
- Clinical + biopsy (tissue confirmation strongly recommended)
- LANA immunostaining for HHV-8 confirmation
- CD4 count and HIV viral load (AIDS-KS)
- CT chest/abdomen/pelvis for staging
- Endoscopy if GI involvement suspected
- Chest X-ray / bronchoscopy if pulmonary KS suspected
Differential diagnosis: Bacillary angiomatosis (can mimic KS — biopsy essential), dermatofibroma, haemangioma, pyogenic granuloma, lichen planus, sarcoidosis
TREATMENT
AIDS-Related KS:
| Severity | Treatment |
|---|
| Mild/Moderate (skin/LN only) | ART alone (most cases respond; immune reconstitution clears KS) |
| Severe/Symptomatic (visceral, oedema, oral interference) | ART + Systemic Chemotherapy |
Systemic Chemotherapy:
- Liposomal doxorubicin (Doxil/Caelyx) — first-line; superior response rate and safety
- Paclitaxel — second-line
- Bleomycin + vincristine — resource-limited settings
Local Therapies (adjunctive):
- Radiotherapy — excellent response; palliation of large lesions, lymphoedema
- Intralesional vinblastine — cosmetically accessible lesions
- Topical alitretinoin gel (9-cis-retinoic acid) — cutaneous KS
- Cryotherapy — small superficial lesions
- Imiquimod — topical immunomodulator
Classic KS:
- Localised: Radiotherapy, surgical excision, cryotherapy, laser
- Extensive: Low-dose systemic chemotherapy (vinblastine, bleomycin)
- Reducing immunosuppression (transplant-associated KS)
Experimental/Targeted:
- Pomalidomide (immunomodulatory)
- Imatinib (KIT inhibitor)
- Anti-VEGF agents
PROGNOSIS
- AIDS-KS: dramatically improved with ART; CD4 recovery is the most important prognostic factor
- Classic KS: indolent; rarely fatal from KS itself
- Lymphadenopathic African KS in children: aggressive; high mortality
- Poor prognostic factors: visceral involvement, CD4 <100, B symptoms
Source: Fitzpatrick's Dermatology 9e (Ch. 168)
E) KLIPPEL-TRENAUNAY-WEBER SYNDROME
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Klippel-Trenaunay Syndrome (KTS) is a sporadic congenital vascular malformation syndrome characterised by the classic triad of:
- Capillary malformation (nevus flammeus / port wine stain)
- Venous and/or lymphatic malformations (varicosities)
- Soft tissue and bony hypertrophy of the affected limb
When an arteriovenous (AV) fistula is additionally present, it is called Klippel-Trenaunay-Parkes-Weber Syndrome (KTPWS).
First described by Klippel and Trenaunay in 1900; Parkes Weber added the AV fistula component.
AETIOLOGY AND PATHOGENESIS
- Sporadic (non-hereditary) in most cases; rarely familial
- Somatic mosaic mutations in PIK3CA (PI3K-AKT-mTOR pathway) — most identified in recent years
- Leads to dysregulated vascular and mesenchymal proliferation during embryogenesis
- Results in failure of normal regression of embryonic lateral vein of the leg (persistent lateral embryonic vein — "marginal vein of Servelle")
- The deep venous system may be hypoplastic or absent (important surgical consideration)
CLINICAL FEATURES
Triad:
1. Capillary Malformation (Port Wine Stain):
- Most common and earliest presenting sign
- Geometric, unilateral, violaceous patch — "geographic" pattern
- Typically involving one limb (lower limb in 95% of cases)
- Does not regress spontaneously; darkens with age
2. Venous and Lymphatic Malformations:
- Lateral varicosities from birth or appearing in childhood
- Persistent lateral embryonic vein (marginal vein) — pathognomonic finding
- Deep venous system may be hypoplastic → contraindication to surgery on superficial veins
- Lymphatic malformations → lymphoedema
- Venous thromboembolism (VTE) in up to 22%
- Venous ulcers, thrombophlebitis
3. Soft Tissue and Bony Hypertrophy:
- Affected limb is larger and longer than normal
- Results from increased blood flow and growth factor stimulation
- Leads to limb length discrepancy and gait abnormalities
- Can affect digits (macrodactyly), causing functional impairment
Additional Features:
- Hypertrichosis over affected limb
- Lymphoedema
- Recurrent cellulitis
- Altered sweating
- Pain, intermittent claudication
- Intradural spinal cord AVMs (at same segmental level) — rare but serious
- Haematuria (genitourinary involvement)
- GI bleeding (visceral haemangiomas — portal hypertension)
- Coagulopathy (Kasabach-Merritt phenomenon in extensive lesions)
Parkes-Weber Syndrome (additional features):
- AV fistula present → warm, pulsatile limb
- High-output cardiac failure risk
- More aggressive hypertrophy
INVESTIGATIONS
| Investigation | Purpose |
|---|
| Colour duplex ultrasonography | Evaluate deep venous system patency; identify AV fistulas |
| MRI with contrast | Visualise extent of soft tissue hypertrophy, vascular malformations |
| Arteriography / Angiography | When AV fistula suspected (Parkes-Weber) |
| Conventional radiography | Both limbs for bony length discrepancy |
| Venography | If deep veins patent → guides surgical planning |
| CT/MRI whole spine | Rule out spinal AVMs |
| Blood coagulation screen | Rule out DIC/Kasabach-Merritt |
DIFFERENTIAL DIAGNOSIS
- Sturge-Weber Syndrome — port wine stain on face (V1 dermatome) + leptomeningeal angioma; no limb hypertrophy
- Proteus Syndrome — progressive asymmetric overgrowth; epidermal naevi; cerebriform connective tissue naevus
- CLOVES Syndrome — PIK3CA mosaic; lipomatous overgrowth + vascular malformations
- Maffucci Syndrome — haemangiomas + enchondromatosis
MANAGEMENT
Multidisciplinary Team: Dermatology + Vascular Surgery + Orthopaedics + Radiology + Haematology
| Problem | Treatment |
|---|
| Port wine stain | Flashlamp pulsed dye laser (PDL, 585/595 nm) — treatment of choice; multiple sessions required |
| Varicosities | Microfoam sclerotherapy; endovenous thermal ablation (EVTA); surgical stripping (only if deep veins patent) |
| Lymphoedema | Elevation; graduated compression garments; graded compression pumps; manual lymphatic drainage; diuretics |
| Limb length discrepancy | Orthopaedic correction — epiphysiodesis (growth plate stapling) in childhood; limb lengthening/shortening procedures |
| AV fistula (PW) | Embolisation; surgical ligation |
| VTE | Anticoagulation (LMWH → warfarin); compression |
| Skin ulcers | Wound care; sunitinib (case reports) |
| Targeted therapy | Sirolimus (rapamycin) — mTOR inhibitor; used for complex vascular malformations; emerging evidence for PIK3CA-mutated cases |
Conservative Measures:
- Compression stockings (life-long)
- Avoid prolonged standing
- Physiotherapy
PROGNOSIS
- Chronic, progressive condition — no cure
- QoL significantly impacted by limb discrepancy and venous complications
- VTE and pulmonary embolism are major causes of mortality
- Regular follow-up for orthopaedic, haematological, and renal complications is mandatory
Source: Andrews' Diseases of the Skin; Fitzpatrick's Dermatology 9e
F) BENIGN ADNEXAL TUMOURS
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Benign adnexal tumours are benign neoplasms arising from the skin appendages — hair follicle, sebaceous glands, eccrine (sweat) glands, and apocrine glands.
CLASSIFICATION
I. FOLLICULAR (Hair Follicle) Origin
| Tumour | Differentiation | Key Features |
|---|
| Trichoepithelioma | Hair germ | Firm, skin-coloured papules on face (nasolabial folds); multiple form is AD (CYLD gene); associated with Brooke-Spiegler syndrome |
| Trichofolliculoma | Follicular hamartoma | Central pore with white "wool-like" hairs protruding; usually solitary on face |
| Trichodiscoma | Fibrous sheaths of follicle | Multiple fibrous papules on nose; Birt-Hogg-Dubé syndrome |
| Pilomatrixoma (Calcifying Epithelioma of Malherbe) | Hair matrix | Solitary, hard, "tent sign" (skin tented over firm mass); calcification; children/young adults; cheek/arm |
| Tricholemmoma | Outer root sheath | Verrucous papule on face; multiple → Cowden syndrome (PTEN mutation) |
| Proliferating Trichilemmal Cyst | Outer root sheath | Scalp; can transform to squamous cell carcinoma |
| Fibrofolliculoma | Mantle of follicle | Multiple skin-coloured papules; Birt-Hogg-Dubé syndrome |
| Dilated Pore of Winer | Infundibulum | Large solitary open comedone; usually face |
II. SEBACEOUS GLAND Origin
| Tumour | Key Features |
|---|
| Sebaceous Hyperplasia | Multiple small yellowish umbilicated papules; forehead/nose; elderly; minocycline useful |
| Sebaceous Adenoma | Yellow lobular tumour; face; multiple → Muir-Torre syndrome (MSH2/MLH1 mutations — associated with visceral malignancy) |
| Sebaceoma | Incompletely differentiated sebaceous tumour |
| Naevus Sebaceus of Jadassohn | Linear/plaque; scalp/face; present at birth; hairless; thickens at puberty; 20% risk of secondary tumours (most commonly trichoblastoma; rarely BCC); excision before puberty recommended |
III. ECCRINE (Sweat Gland) Origin
| Tumour | Key Features |
|---|
| Eccrine Hidrocystoma | Solitary translucent cyst; periorbital (lower eyelid); increases in summer heat; needle drainage curative |
| Syringoma | Multiple, skin-coloured, small firm papules; lower eyelids bilaterally; young women; ducts within fibrous stroma on histology; associated with Down syndrome |
| Eccrine Poroma | Solitary reddish vascular papule/plaque on sole; "stuck-on"; origin from acrosyringium |
| Chondroid Syringoma (Mixed Tumour) | Firm nodule; face/scalp; ducts in chondromyxoid stroma; resembles pleomorphic adenoma of salivary gland |
| Eccrine Spiradenoma | Solitary painful nodule (spontaneous pain); trunk; young adults; painful skin tumours mnemonic: ANGEL — Angiolipoma, Neuroma, Glomus, Eccrine spiradenoma, Leiomyoma |
| Cylindroma (Turban Tumour) | Multiple coalescing nodules on scalp (resembles turban); AD; Brooke-Spiegler syndrome (CYLD gene) |
| Milia | Tiny white keratin cysts; face; secondary to bullous disorders or trauma |
IV. APOCRINE GLAND Origin
| Tumour | Key Features |
|---|
| Apocrine Hidrocystoma | Translucent bluish cyst; ear, periorbital; larger than eccrine type; contents smell rancid |
| Syringocystadenoma Papilliferum | Linear warty plaque; scalp; may arise in naevus sebaceus; plasma cell infiltrate on histology |
| Hidradenoma Papilliferum | Vulva/perineum of women; papillary projections on histology |
| Apocrine Adenoma | Axilla, anogenital region |
HISTOPATHOLOGICAL PATTERNS (Key for Exams)
| Pattern | Tumour Type |
|---|
| "Puzzle piece" or jigsaw basaloid islands | Trichoepithelioma (vs BCC — no ulceration, no mucin, papillary stroma) |
| Shadow/ghost cells with calcification | Pilomatrixoma |
| Bilayered ductal structures in fibrous stroma | Syringoma |
| Tadpole-shaped cells, acrosyringial differentiation | Eccrine poroma |
| Hyaline cylinder around cell islands ("cannon ball") | Cylindroma |
| Papillary projections with plasma cells | Syringocystadenoma papilliferum |
IMPORTANT SYNDROME ASSOCIATIONS
| Syndrome | Tumour | Gene | Associated Malignancy |
|---|
| Brooke-Spiegler | Cylindroma, trichoepithelioma, spiradenoma | CYLD | Rare malignant transformation |
| Muir-Torre | Sebaceous adenoma/carcinoma | MSH2, MLH1 | Colorectal, GU cancers |
| Cowden | Multiple trichilemmomas | PTEN | Breast, thyroid, endometrial cancer |
| Birt-Hogg-Dubé | Fibrofolliculoma, trichodiscoma | FLCN (folliculin) | Renal cell carcinoma, lung cysts |
| Gardner Syndrome | Epidermoid cysts, pilomatrixomas, fibromas | APC | Colorectal polyposis/cancer |
TREATMENT
| Modality | Used For |
|---|
| Surgical excision | Most solitary tumours |
| Laser (CO₂, Er:YAG) | Multiple facial lesions (syringomas, trichoepitheliomas) |
| Dermabrasion | Multiple small lesions |
| Electrodesiccation | Small superficial lesions |
| Topical retinoids | Syringoma (partial response) |
| Oral retinoids | Extensive/syndromic cases |
| Genetic counselling | Syndromic cases (Muir-Torre, Cowden, BHD) |
Sources: Fitzpatrick's Dermatology 9e; Andrews' Diseases of the Skin
G) LEUKOPLAKIA
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Leukoplakia (WHO, 2005) is defined as "a white patch or plaque of the oral mucosa that cannot be characterised clinically or pathologically as any other disease" and which cannot be removed by scraping. It is a potentially malignant disorder (PMD).
EPIDEMIOLOGY
- Prevalence: 0.2–4.9% globally
- Male predominance (M:F = 2–3:1)
- Most common in: South and Southeast Asia (tobacco + betel nut use)
- Malignant transformation rate: 0.7–2.9% per year (overall ~5–17%)
AETIOLOGY / RISK FACTORS
| Factor | Notes |
|---|
| Tobacco (most important) | Smoking + smokeless (chewing, betel quid); dose-dependent risk |
| Alcohol | Synergistic with tobacco |
| Betel nut / areca nut | South Asia; high-risk combination (pan, gutka, khaini) |
| HPV (esp. types 16, 18) | Especially in proliferative verrucous leukoplakia |
| Candida | Candida-associated leukoplakia; increased malignant potential |
| Chronic trauma | Ill-fitting dentures, sharp teeth |
| Nutritional | Iron deficiency, vitamin A and B12 deficiency |
| Syphilis | Syphilitic glossitis; high malignant potential |
CLASSIFICATION
A. WHO Classification (Pindborg, 1997 / updated):
| Type | Features | Malignant Risk |
|---|
| Homogeneous | Uniformly white, flat/slightly raised, smooth or wrinkled surface; sharp margins | Low (1–7%) |
| Non-homogeneous | Irregular surface; further subdivided: | Higher |
| → Erythroleukoplakia (Speckled) | White patches on red background (erythroplakia) | High (>20%) |
| → Nodular | Small rounded white/red excrescences | High |
| → Verrucous | Irregular, wrinkled, corrugated surface | High |
| Proliferative Verrucous Leukoplakia (PVL) | Multifocal; progressive; elderly women; HPV-associated; high recurrence | Very high (60–100%) |
B. Based on Size:
- L1: <2 cm; L2: 2–4 cm; L3: >4 cm (size correlates with malignant risk)
SITES AND MALIGNANT POTENTIAL
| Site | Malignant Risk |
|---|
| Floor of mouth | Highest |
| Ventral tongue | Very high |
| Soft palate, tonsillar region | High |
| Buccal mucosa (most common site overall) | Low-intermediate |
| Hard palate | Low |
| Labial commissure | Low |
CLINICAL FEATURES
- Homogeneous leukoplakia: Smooth, uniformly white, flat or slightly raised plaque; well-defined borders; often asymptomatic; usually buccal mucosa or alveolar ridge
- Non-homogeneous: Irregular, may have red components; more likely to be dysplastic
- May present on: buccal mucosa (most common), gingiva, tongue (lateral border and ventral surface = high risk)
- Oral hairy leukoplakia (OHL): White, corrugated "hairy" patches on lateral border of tongue; non-removable; caused by EBV replication; marker of HIV immunosuppression (not truly pre-malignant)
HISTOPATHOLOGY
Epithelial dysplasia grading is the most important histological finding:
| Grade | Features |
|---|
| Mild dysplasia | Atypia in lower third of epithelium |
| Moderate dysplasia | Atypia in lower two-thirds |
| Severe dysplasia | Atypia throughout (near-carcinoma in situ) |
| Carcinoma in situ | Full-thickness atypia; no invasion |
Dysplastic features: Nuclear pleomorphism, increased N:C ratio, abnormal mitoses (suprabasal), dyskeratosis, drop-shaped rete ridges, loss of polarity, irregular epithelial stratification
MALIGNANT POTENTIAL — HIGH-RISK INDICATORS
| Feature | High Risk |
|---|
| Site | Floor of mouth, ventral/lateral tongue, soft palate |
| Type | Non-homogeneous, erythroleukoplakia, PVL |
| Size | >200 mm² |
| Histology | Moderate–severe dysplasia or CIS |
| Habits | Non-tobacco user (paradoxically higher risk) |
| Gender | Female (higher risk) |
| Duration | Long-standing |
| Candida | Positive |
DIAGNOSIS
- Clinical examination + history
- Toluidine blue staining — screens for dysplasia (stains DNA in dividing cells; high sensitivity, moderate specificity)
- Biopsy (incisional, from most suspicious area) — mandatory for all leukoplakias
- VELscope / Autofluorescence — identifies dysplastic areas
- Smear for Candida (PAS stain)
- HPV testing (emerging)
DIFFERENTIAL DIAGNOSIS
| Condition | Distinguishing Feature |
|---|
| Oral Candidiasis (Thrush) | White plaques that can be scraped off (unlike leukoplakia) |
| Lichen Planus | Wickham's striae; bilateral; violaceous papules elsewhere |
| Oral Hairy Leukoplakia | Lateral tongue; HIV; EBV; cannot be scraped; corrugated |
| Linea Alba | Thin white line along occlusal plane; physiological |
| Fordyce Spots | Ectopic sebaceous glands; yellowish |
| White Sponge Naevus | Familial; large, bilateral, benign |
| Erythroplakia | Red velvety plaque; >90% dysplasia/carcinoma |
MANAGEMENT
I. Eliminate Risk Factors:
- Stop tobacco, alcohol, betel nut — most important step
- Remove local irritants (dentures, sharp cusps)
- Treat Candida (fluconazole)
II. Observation (with regular follow-up):
- Homogeneous leukoplakia without dysplasia; <2 cm; easily accessible
- 3–6 monthly reviews; re-biopsy if changes
III. Treatment of Dysplastic/High-risk Lesions:
| Method | Indication |
|---|
| Surgical excision (scalpel) | Gold standard for all dysplastic lesions; provides histological specimen |
| CO₂ laser excision/ablation | First choice for extensive/multifocal lesions; good cosmetic outcome |
| Cryotherapy | Small accessible lesions |
| Photodynamic therapy (PDT) | Multifocal/extensive dysplastic leukoplakia; emerging |
IV. Medical/Chemoprevention:
| Drug | Evidence |
|---|
| Topical bleomycin | Reduces lesion size; partial regression |
| Retinoids (isotretinoin, acitretin) | Partial response; high relapse on stopping |
| Beta-carotene | Antioxidant; modest benefit |
| Lycopene | Antioxidant; some evidence |
| Topical podophyllin | Outdated; not recommended |
V. Transformation to SCC:
- Surgical resection with clear margins ± neck dissection
- Radiotherapy, chemotherapy per oncology protocol
PROGNOSIS AND FOLLOW-UP
- All patients need life-long surveillance (6–12 monthly)
- Recurrence after treatment: up to 30% for surgical excision; higher for laser/cryotherapy
- PVL has very high recurrence and transformation rate
Sources: K.J. Lee's Essential Otolaryngology; Cummings Otolaryngology; Fitzpatrick's Dermatology 9e
H) STAPHYLOCOCCAL SCALDED SKIN SYNDROME (SSSS)
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
Staphylococcal Scalded Skin Syndrome (SSSS) is a generalised, superficially exfoliative disease caused by the distant effects of exfoliative exotoxins (ETs) A and B produced by Staphylococcus aureus at a remote site of infection. The skin exfoliates in sheets, resembling a thermal scald.
EPIDEMIOLOGY
- Primarily affects neonates and children under 5 years
- Rare in adults (predisposing factors: renal failure, immunosuppression, HIV)
- In adults, renal clearance of toxin is normally rapid — absence of renal function → toxin accumulates
- Causative organism: Group 2 S. aureus, phage types 71 or 55 (most common)
- MSSA is more common than MRSA in SSSS
AETIOLOGY AND PATHOGENESIS
Mechanism:
- S. aureus colonises/infects a remote site (nose, pharynx, ear, conjunctiva, umbilicus in neonates, urinary tract)
- Produces exfoliative toxins A and B (ET-A, ET-B) — serine proteases
- Toxins are absorbed systemically and travel to the skin
- ET-A and ET-B specifically cleave Desmoglein 1 (Dsg-1) — the same target as pemphigus foliaceus autoantibodies
- Dsg-1 is the predominant adhesion molecule in the superficial epidermis (granular layer)
- Cleavage of Dsg-1 → split in the granular layer (subcorneal) → superficial blister
- Dsg-3 (predominant in mucosae and deep epidermis) is not cleaved → mucous membranes spared
Why children are susceptible:
- Lack of protective antibodies against ET
- Immature renal clearance → higher circulating toxin levels
- Smaller surface area:volume ratio
CLINICAL FEATURES
Stages:
Stage 1 — Prodrome:
- Fever, irritability, skin tenderness
- Diffuse erythema with perioral, periorbital, and intertriginous accentuation (neck, groin, axillae)
- Skin described as resembling a "sunburn"; painful to touch
Stage 2 — Exfoliative:
- Nikolsky sign positive — lateral pressure on erythematous skin → epidermal sliding/slipping
- Blisters form at sites of pressure (where ECG leads are attached/removed; where infant is picked up)
- Large, flaccid bullae → rupture → leave denuded, glistening, "scalded" skin
- Exfoliation in large sheets → begins perioral and spreads centrally
Stage 3 — Desquamation:
- Widespread peeling with moist, red, denuded areas
- Crusting and desquamation in 3–5 days
- Re-epithelialisation within 1–2 weeks (lesions are very superficial)
Key Distinguishing Features:
- Mucous membranes SPARED (unlike SJS/TEN)
- Palms and soles spared
- Skin is sterile (unlike bullous impetigo — S. aureus not in lesion, only at remote source)
- Patient appears in pain, irritable
COMPARISON: SSSS vs TEN vs BULLOUS IMPETIGO
| Feature | SSSS | TEN | Bullous Impetigo |
|---|
| Age | Neonates, <5 years | Any age | Any (mainly children) |
| Cause | S. aureus ETs (remote site) | Drug | S. aureus (local) |
| Level of split | Subcorneal/granular layer | Dermoepidermal junction (full thickness) | Subcorneal |
| Mucous membranes | Spared | Involved | Spared |
| Nikolsky sign | Positive | Positive | Negative (localised) |
| Skin culture | Negative (sterile lesions) | Negative | Positive |
| Healing | Rapid (1–2 weeks) | Slow; scarring | Rapid |
| Mortality (untreated) | Low in children; higher in adults | High (30–50% for TEN) | Very low |
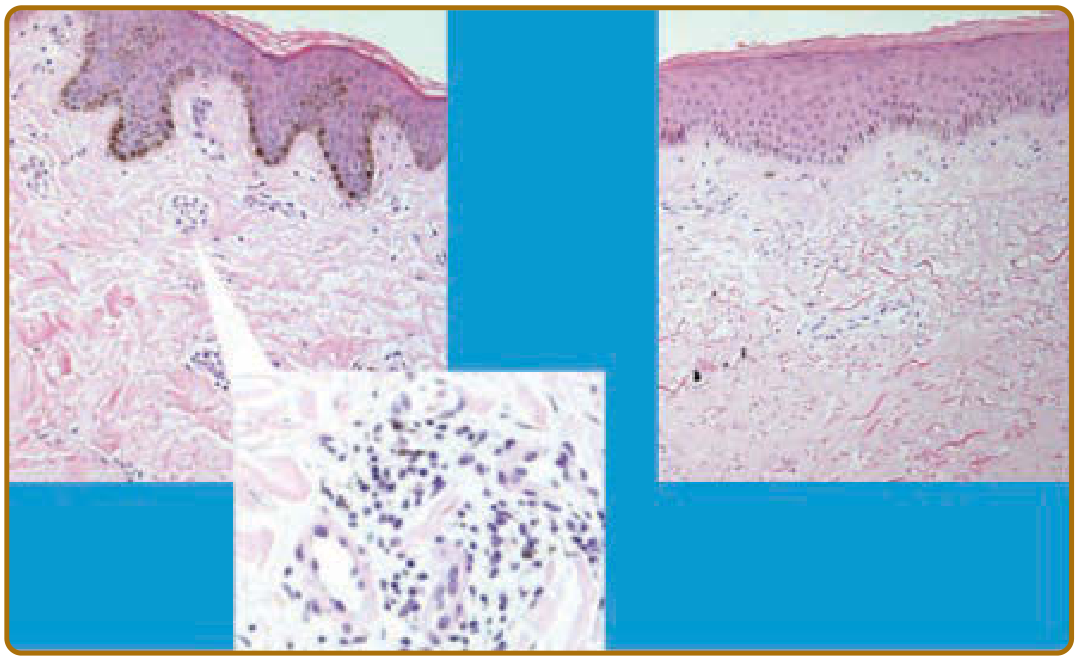
HISTOPATHOLOGY
- Split in the granular layer (subcorneal) — cleavage plane is just below stratum corneum
- Full-thickness epidermal necrosis is absent (cf. TEN)
- Superficial acantholysis; no significant inflammation
Frozen section of blister roof: Quick diagnosis — full-thickness epidermis in TEN vs only superficial corneum in SSSS.
DIAGNOSIS
Primarily clinical. Confirm with:
- Cultures from remote focus (nares, throat, conjunctivae, perianal area, umbilicus, blood, urine)
- Skin biopsy / frozen section of blister roof — shows subcorneal split
- FBC: leukocytosis
- Urea, creatinine: assess renal function
- Blood culture: if bacteraemia/septicaemia suspected
MANAGEMENT
Antibiotics (Definitive Treatment):
| Antibiotic | Indication |
|---|
| Penicillinase-resistant penicillin — Cloxacillin/Dicloxacillin | First-line for MSSA (most common) |
| IV Flucloxacillin | Severe/systemic disease; neonates; hospitalised patients |
| Clindamycin (+ β-lactam) | Inhibits ribosomal toxin synthesis; reduces ET production; useful adjunct — but use only with susceptibility confirmed |
| Vancomycin / Linezolid | If MRSA confirmed on cultures |
| Duration: 5–10 days | |
Note: Clindamycin resistance is increasing — always use culture-guided therapy.
Supportive Care:
| Component | Details |
|---|
| Fluid replacement | Oral or IV; replace insensible fluid losses from denuded skin |
| Temperature regulation | Warm environment; prevent hypothermia |
| Skin care | Gentle non-adherent dressings; petroleum gauze; avoid harsh soaps |
| Analgesia | Paracetamol; avoid NSAIDs |
| Nutritional support | Nasogastric feeds in severe neonatal cases |
| Ophthalmology | Not typically required (mucosae spared), but assess if periocular involvement |
Monitoring:
- Fluid balance (denuded skin = insensible losses similar to burns)
- Watch for secondary infection
- In adults with renal failure: dialysis may be needed (to clear circulating toxin)
PROGNOSIS
- In children: Excellent — rapid healing within 1–2 weeks; no scarring
- In adults (especially with renal failure/immunosuppression): Mortality up to 60% without treatment
- No post-inflammatory scarring as split is superficial (cf. TEN — scarring and sequelae)
Source: Andrews' Diseases of the Skin; Goldman-Cecil Medicine
I) CLASSIFY ICHTHYOSIS; ETIOPATHOGENESIS AND CLINICAL FEATURES OF CONGENITAL ICHTHYOSIS
3rd Year Dermatology PG Theory — 10 Marks
DEFINITION
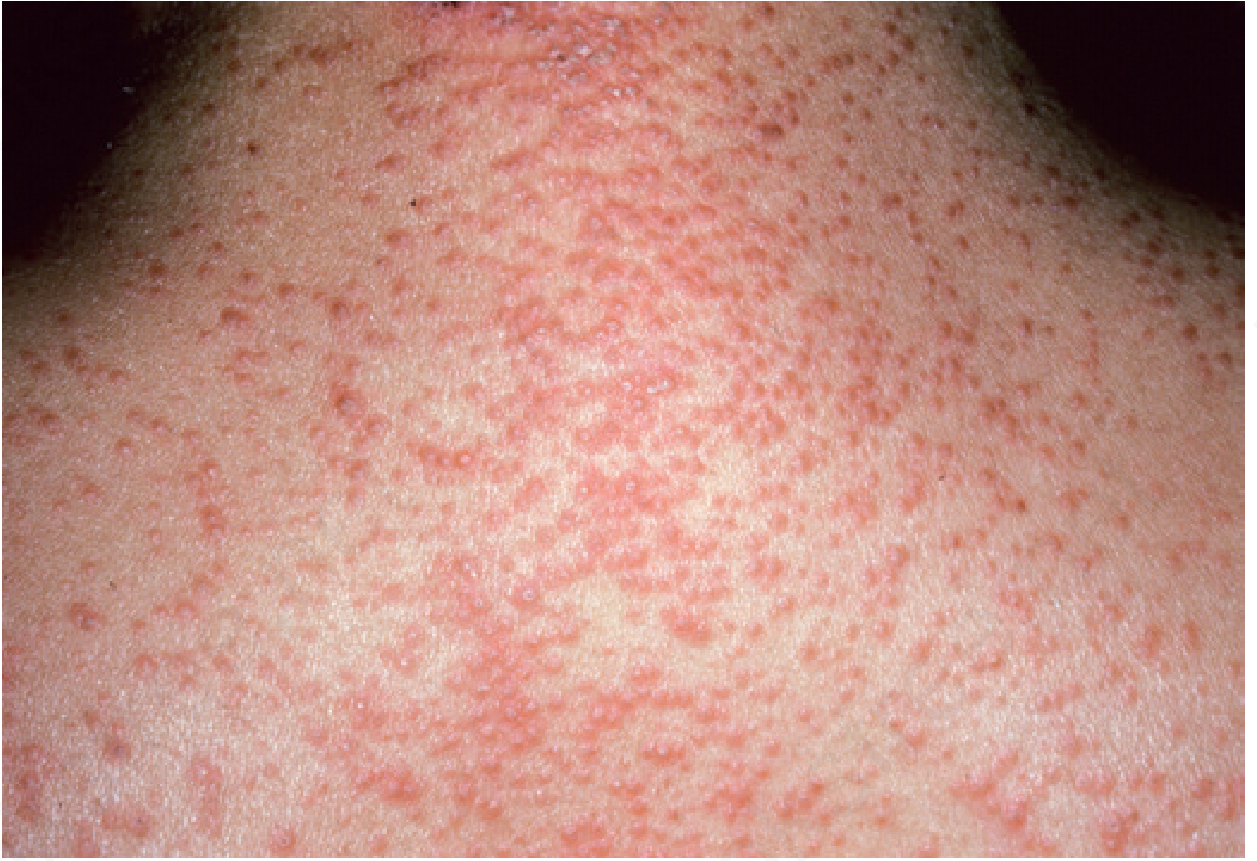
Ichthyosis is a group of inherited or acquired disorders of keratinisation characterised by generalised scaling of the skin, resembling fish scales (Greek: ichthys = fish). It results from either increased epidermal proliferation or decreased desquamation of the stratum corneum.
CLASSIFICATION
I. INHERITED ICHTHYOSES
A. Common Ichthyoses:
| Type | Inheritance | Gene | Incidence |
|---|
| Ichthyosis Vulgaris (IV) | Semidominant | FLG (filaggrin) | 1:80–1:250 (most common) |
| X-Linked Recessive Ichthyosis (XLRI) | X-linked recessive | STS (steroid sulphatase) | 1:2000–6000 males |
B. Autosomal Recessive Congenital Ichthyoses (ARCI) — formerly "Non-bullous CIE":
| Phenotype | Gene(s) | Notes |
|---|
| Lamellar Ichthyosis (LI) | TGM1 (most common), ABCA12, CYP4F22, NIPAL4 | Large, dark, plate-like scales; collodion baby |
| Congenital Ichthyosiform Erythroderma (CIE) | ALOX12B, ALOXE3, TGM1 | Fine white scales on erythrodermic base |
| Harlequin Ichthyosis (HI) | ABCA12 (most severe) | Armour-like plates at birth |
| Bathing Suit Ichthyosis | TGM1 (temperature-sensitive) | Scaling limited to bathing suit distribution |
C. Autosomal Dominant Congenital Ichthyoses (ADCI) — "Bullous" forms:
| Type | Gene | Features |
|---|
| Epidermolytic Ichthyosis (EI) (formerly BCIE) | KRT1, KRT10 | Blistering at birth + hyperkeratosis; keratin defect |
| Superficial Epidermolytic Ichthyosis | KRT2 | Milder; bullae less prominent |
D. Syndromic Ichthyoses (Ichthyosis as part of multi-organ syndrome):
| Syndrome | Ichthyosis Type | Associated Features |
|---|
| Netherton Syndrome | Ichthyosis linearis circumflexa (ILC) | SPINK5 gene; trichorrhexis invaginata (bamboo hair); atopy; immune dysregulation |
| Sjögren-Larsson Syndrome | Congenital ichthyosis | ALDH3A2 gene; spastic diplegia; intellectual disability; glistening retinal dots |
| Refsum Disease | Ichthyosis | PHYH/PEX7 gene; phytanic acid accumulation; retinitis pigmentosa; peripheral neuropathy; ataxia |
| CHILD Syndrome | Unilateral ichthyosis | NSDHL/EBP gene; ipsilateral limb defects; X-linked dominant |
| KID Syndrome | Keratitis-Ichthyosis-Deafness | GJB2 (connexin 26); keratitis (blindness risk); neurosensory deafness |
| Trichothiodystrophy (TTD) | Collodion baby → generalised scaling | DNA repair defects (XPD/ERCC2); brittle hair; photosensitivity; intellectual disability |
II. ACQUIRED ICHTHYOSIS
| Cause | Notes |
|---|
| Malignancy (most important) | Hodgkin's lymphoma (classic), non-Hodgkin's, multiple myeloma |
| Drugs | Nicotinic acid, triparanol, butyrophenones, cimetidine |
| Metabolic | Hypothyroidism, malnutrition, malabsorption, renal failure |
| Infection | HIV (rare), leprosy |
| Autoimmune | SLE, dermatomyositis, sarcoidosis |
ETIOPATHOGENESIS OF CONGENITAL ICHTHYOSIS
Normal Epidermal Barrier:
- Keratinocytes proliferate in the basal layer → differentiate → cornify → desquamate
- The stratum corneum (SC) = "brick and mortar" structure: corneocytes (bricks) + lipid lamellae (mortar)
- Controlled desquamation by serine proteases (kallikreins) and their inhibitors (LEKTI/SPINK5)
Pathogenetic Mechanisms:
1. Defective Cornified Envelope / Structural Protein Defects
Ichthyosis Vulgaris (FLG mutation):
- Filaggrin (from profilaggrin) aggregates keratin intermediate filaments and forms the cornified cell envelope
- Loss-of-function mutations → absent/reduced filaggrin → inadequate corneocyte integrity → xerosis and scaling
- Also disrupts natural moisturising factor (NMF) production (filaggrin breakdown products) → impaired barrier → ↑ TEWL
- Semidominant: heterozygotes = mild IV + atopy risk; homozygotes = severe IV
- Associated with atopic dermatitis (50% of IV patients have atopy)
2. Defective Lipid Metabolism/Transport
XLRI (Steroid Sulphatase deficiency):
- STS enzyme cleaves cholesterol sulphate in SC
- Deficiency → accumulation of cholesterol sulphate → inhibits serine proteases → impaired desquamation
- Scaling is due to retention hyperkeratosis (scale retained, not shed)
ABCA12 (Harlequin/Lamellar Ichthyosis):
- ABCA12 is a lipid transporter in lamellar granules (Odland bodies)
- Deficiency → failure to deliver lipid lamellae into intercellular space of SC → severely defective barrier
- Most severe mutation: Harlequin ichthyosis (complete loss of ABCA12)
ARCI — Lipoxygenase defects (ALOX12B, ALOXE3):
- These enzymes process polyunsaturated fatty acids for epidermal ceramide synthesis
- Deficiency → defective ceramide production → abnormal lipid lamellae → impaired barrier → scaling
3. Defective Transglutaminase — Cross-linking Defect
TGM1 (Lamellar Ichthyosis):
- Transglutaminase-1 cross-links proteins of the cornified cell envelope
- Deficiency → structurally defective corneocytes → abnormal cohesion → scaling
- Bathing suit ichthyosis: temperature-sensitive TGM1 mutations — reduced activity at higher temperatures (trunk) → limited distribution
4. Defective Keratin Filaments — Cytoskeletal Defect
Epidermolytic Ichthyosis (KRT1/KRT10 mutations):
- KRT1 and KRT10 are the main keratins of suprabasal keratinocytes
- Mutations → keratin tonofilament collapse → cytolysis → blistering
- Blister healing → abnormal hyperkeratosis (thickened epidermis as repair response)
- Histology: epidermolytic hyperkeratosis — suprabasal cytolysis with shell of condensed keratin
5. Defective Protease Inhibition — Netherton Syndrome
- SPINK5 encodes LEKTI (Lympho-Epithelial Kazal-type related Inhibitor) — inhibitor of skin kallikreins
- Loss of LEKTI → uninhibited kallikrein 5/7 activity → excessive desquamation + impaired barrier
- Also activates PAR-2 → Th2 inflammation → atopy (eczema, high IgE, food allergies)
- Results in ichthyosis linearis circumflexa (ILC) — polycyclic, serpiginous migrating plaques
CLINICAL FEATURES OF CONGENITAL ICHTHYOSIS (ARCI)
A. COLLODION BABY — Common Presenting Phenotype of ARCI
A collodion baby is a neonate presenting with a taut, shiny, parchment-like membrane (resembling collodion — a film-forming solution) encasing the entire body.
Features:
- Restricted mouth opening (feeding difficulty) and chest movement (respiratory compromise)
- Ectropion (eversion of eyelids) and eclabium (eversion of lips)
- Fissuring at flexures (neck, elbows, knees)
- Limbs rigid, fingers taped together; ears filled with keratin
- Management: Humidified incubator; emollients (liquid paraffin); NG tube feeding; eye care (lubricating drops)
Outcomes of collodion baby:
- Lamellar ichthyosis (~30%)
- CIE (~30%)
- Self-healing collodion baby (SHCB) — rare; resolves completely
- Normal skin (~10%)
- Other ichthyoses
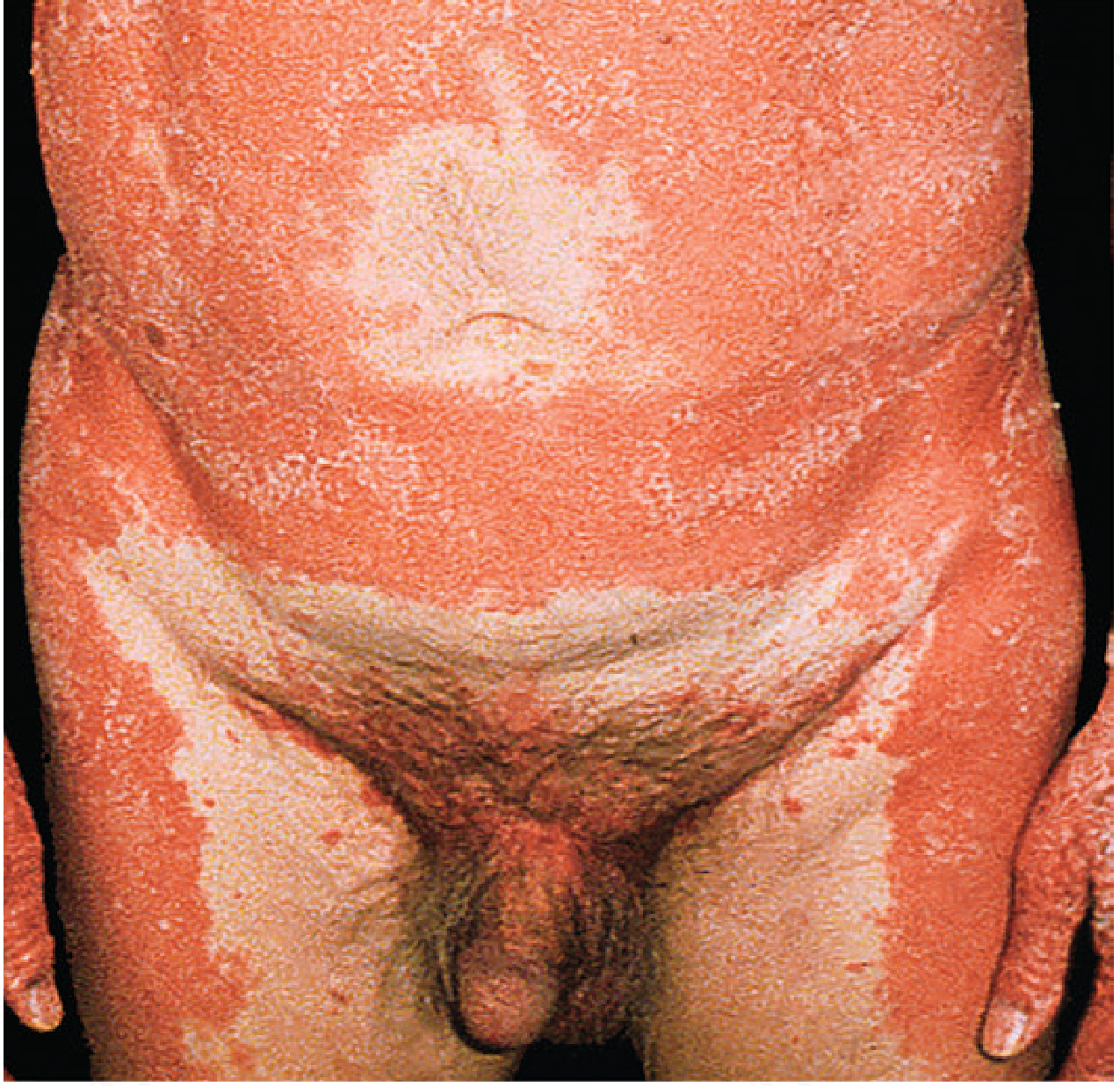
B. LAMELLAR ICHTHYOSIS (LI)
Gene: TGM1 (most common), ABCA12, CYP4F22, etc.
Clinical Features:
- Presents at birth as collodion baby
- After membrane shedding: large, plate-like, dark, centrally adherent scales with raised borders
- Scales largest over lower extremities ("dry riverbed" appearance)
- Erythema variable (may be mild or absent)
- Ectropion (eversion of lower eyelids — important complication; risk of corneal damage)
- Eclabium (eversion of lips)
- Scarring alopecia at scalp periphery (scales entrap and break hairs)
- Hypohidrosis (scales block eccrine ducts) → heat intolerance; serious risk of hyperthermia
- Palmoplantar keratoderma (variable — minimal hyperlinearity to severe)
- Nails: thickened, ridged
- Mucous membranes and lips usually spared
- Dermatophyte infections of skin and nails are common
C. CONGENITAL ICHTHYOSIFORM ERYTHRODERMA (CIE)
Gene: ALOX12B, ALOXE3, TGM1, NIPAL4
Clinical Features:
- Also presents as collodion baby
- After membrane shedding: persistent erythroderma with fine, white, generalised scale
- Lower legs: larger, darker scales
- Less ectropion, eclabium, alopecia than LI (classic CIE)
- Variable sweating impairment → heat intolerance
- Mucous membranes usually spared; nails may be ridged
D. HARLEQUIN ICHTHYOSIS — Most Severe ARCI
Gene: ABCA12 (complete loss of function)
Clinical Features:
- Neonate born with thick, armour-like plates of stratum corneum separated by deep red fissures
- Geometric pattern of fissures (resembling harlequin costume)
- Rudimentary/absent ears (sealed with keratin)
- Profound ectropion and eclabium
- Tapered fingertips; hyperconvex nails
- Restricted chest movement → respiratory failure
- Feeding impossible (mouth restricted)
- Historically uniformly fatal before 1980
- Now: survival possible with early intensive care + oral retinoids (acitretin)
- Survivors have persistent, severe erythroderma resembling CIE
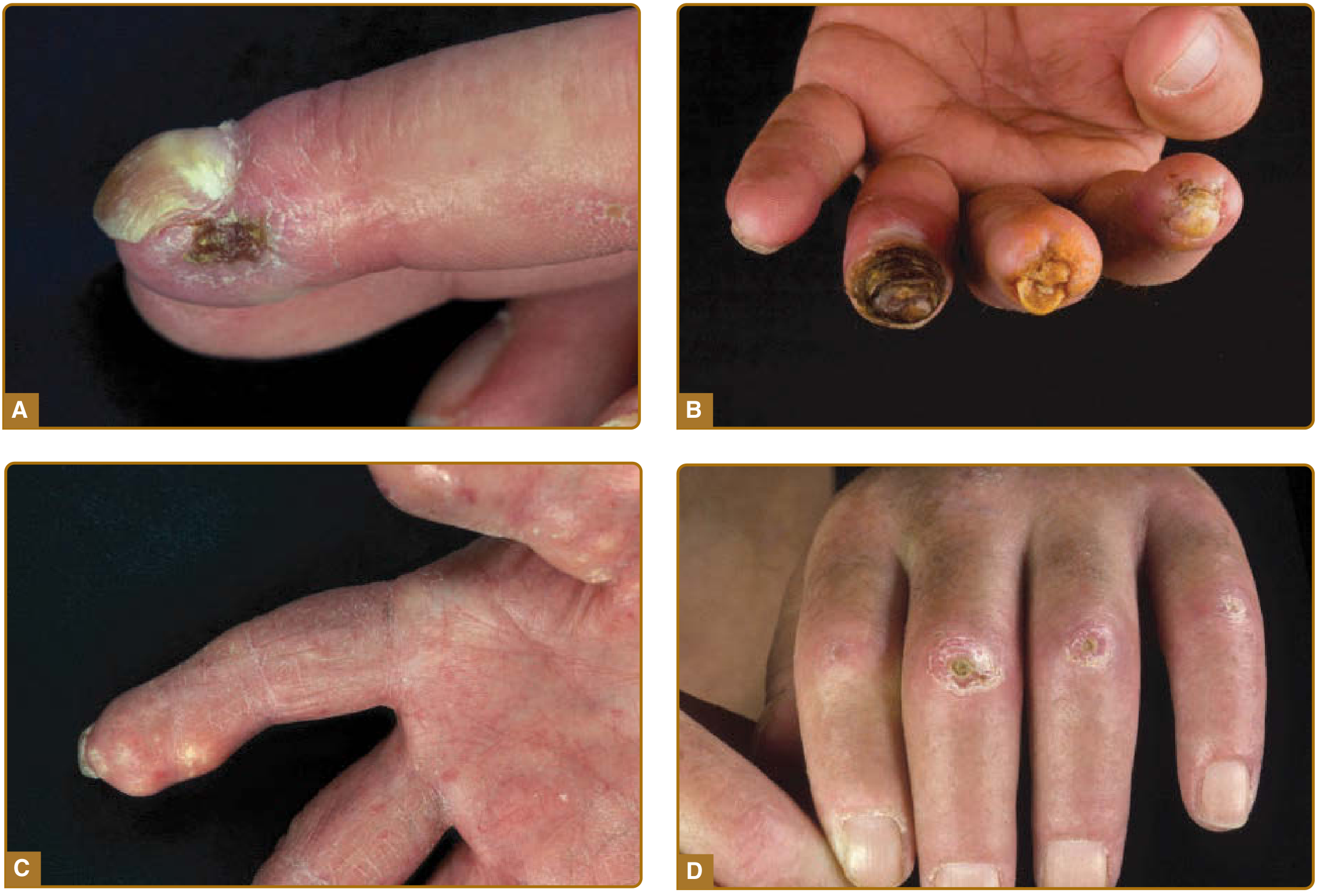
E. EPIDERMOLYTIC ICHTHYOSIS (EI) — ADCI
Gene: KRT1, KRT10 (autosomal dominant)
Clinical Features:
- At birth: generalised blistering + erythroderma; resembles SSSS or EBS
- Bullae heal, leaving hyperkeratosis
- With age: blistering decreases; severe verrucous hyperkeratosis develops (especially flexures — neck, axillae, groin)
- Offensive odour (bacterial colonisation of hyperkeratotic skin)
- Palmar-plantar keratoderma (if KRT1 involved)
- Histology: Epidermolytic hyperkeratosis — suprabasal vacuolisation, keratin clumping, granular layer changes
INVESTIGATION
| Test | Purpose |
|---|
| Skin biopsy + histopathology | Specific patterns per type |
| Electron microscopy | Ultrastructural defects (lamellar granules in HI) |
| Genetic testing (NGS panel) | Definitive diagnosis; guides prognosis |
| Transglutaminase 1 activity (frozen skin) | Reduced/absent in TGM1-related LI |
| Plasma phytanic acid | Refsum disease |
| Hair microscopy | Trichorrhexis invaginata (Netherton), pili torti (TTD) |
TREATMENT
General Measures:
- Emollients (cornerstone of all ichthyoses): urea 5–10%, lactic acid, glycerine; apply multiple times daily
- Keratolytics: Urea 10–20%, salicylic acid 2–6%, lactic acid 5–12% (not in neonates — systemic absorption)
- Bathing: Long daily soaks → mechanical removal of scales; add salt (NaCl) or oil
- Humidified environment (especially HI neonates)
- Temperature control: Air conditioning; avoid exercise in heat (hypohidrosis)
- Treat secondary infections (Candida, dermatophytes, S. aureus)
Systemic Treatment:
| Drug | Indication | Notes |
|---|
| Acitretin | LI, CIE, HI (life-saving), Netherton | Most effective systemic; reduces scale and erythema; long-term use; teratogenic |
| Isotretinoin | EI, syndromic ichthyoses | |
| Statins | XLRI (off-label; reduce cholesterol sulphate) | |
Targeted / Emerging:
- Thymosin β4 (experimental in HI)
- Gene therapy — under investigation for TGM1-related LI
- Enzyme replacement — STS for XLRI (experimental)
Ophthalmology:
- Regular review for ectropion management (lubricating drops, tarsorrhaphy)
Genetic Counselling:
- All families should receive counselling
- Prenatal diagnosis via molecular testing available for known family mutations
Key exam points across all topics:
- Melanoma: Breslow thickness = single most important prognostic factor; acral → KIT; BRAF in SSM → targeted therapy
- Dermatological emergencies: SCORTEN for SJS/TEN; granulysin = key mediator of TEN
- Kaposi sarcoma: HHV-8; LANA IHC; liposomal doxorubicin first-line systemic
- KTS: PIK3CA mutations; persistent marginal vein; check deep veins before surgery; sirolimus for vascular malformations
- Adnexal tumours: Muir-Torre (sebaceous + colorectal Ca); Cowden (trichilemmoma + breast Ca); BHD (fibrofolliculoma + renal Ca)
- Leukoplakia: Dsg-1 cleavage → mucosae spared in SSSS (Dsg-3 predominates there)
- SSSS: Dsg-1 cleavage by ETs; subcorneal split; mucosae spared; sterile lesions; children; flucloxacillin
- Ichthyosis: Collodion baby is a presenting phenotype, not a diagnosis; TGM1 = LI; ABCA12 = Harlequin; FLG = ichthyosis vulgaris; acitretin is the mainstay systemic treatment
Sources: Fitzpatrick's Dermatology 9e; Andrews' Diseases of the Skin; Robbins Pathologic Basis of Disease; Goldman-Cecil Medicine