Explain the mechanism of acute respiratory distress syndrome
Reading File
search_medical_text
acute respiratory distress syndrome pathophysiology mechanism
search_medical_text
ARDS diffuse alveolar damage inflammation cytokines endothelial injury
search_medical_image
ARDS diffuse alveolar damage pathophysiology diagram
GradeResponse
GradeResponse
GradeResponse
Here is a comprehensive breakdown of the mechanism of ARDS.
Acute Respiratory Distress Syndrome (ARDS) — Mechanism
Overview
ARDS is a clinical-pathological syndrome characterized by three core physiological derangements (ARDS: Definition, Phenotyping and Respiratory Support Strategies, p. 2):
- Increased alveolo-capillary membrane permeability → inflammatory pulmonary edema
- Increased non-aerated lung tissue → reduced compliance (high lung elastance)
- Increased venous admixture and dead space → hypoxemia and hypercapnia
The underlying histological pattern is diffuse alveolar damage (DAD), which evolves through three overlapping phases.
The Three Phases of ARDS
Phase 1: Exudative Phase (Days 1–7)
This is the acute inflammatory phase and the core of ARDS pathogenesis (Harrison's Principles of Internal Medicine, 21st ed., p. 8197):
Cellular injury:
- Alveolar capillary endothelial cells and type I pneumocytes are the primary targets of injury
- Their destruction breaks down the normally tight alveolar-capillary barrier
Fluid and protein leak:
- Loss of barrier integrity allows protein-rich edema fluid to flood the interstitial and alveolar spaces
- This impairs gas exchange across the alveolar membrane
Cytokine storm and neutrophil recruitment:
- Proinflammatory cytokines are massively upregulated:
- IL-1, IL-8 — potent neutrophil chemoattractants
- TNF-α — promotes endothelial activation and permeability
- Leukotriene B₄ — lipid mediator amplifying neutrophil influx
- Neutrophils flood the pulmonary interstitium and alveoli, releasing:
- Reactive oxygen species (ROS)
- Myeloperoxidase (MPO)
- Neutrophil extracellular traps (NETs)
- Proteases — all causing further tissue destruction
Hyaline membrane formation:
- Condensed plasma proteins, cellular debris, and dysfunctional surfactant aggregate to form hyaline membrane whorls lining the alveolar walls — a hallmark of DAD on histology
Vascular injury:
- Pulmonary microvascular injury causes microthrombi and fibrocellular proliferation, obliterating the pulmonary vasculature and increasing dead space
Molecular Cascade Diagram
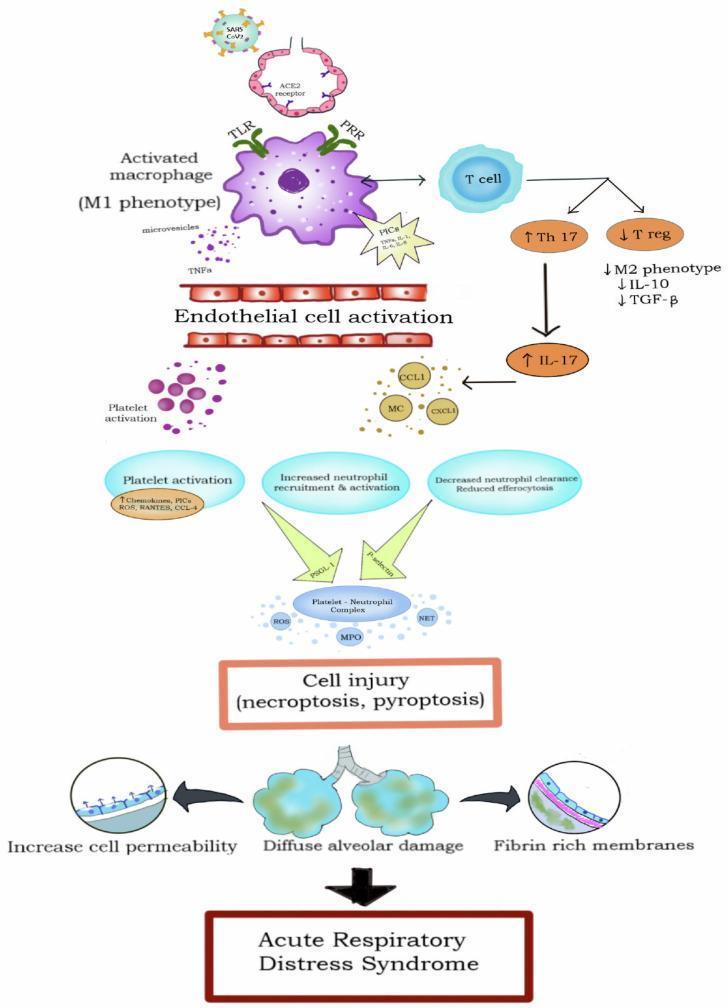
The diagram above shows how viral/infectious triggers activate macrophages (M1 phenotype) via TLR/PRR pathways → Th17 upregulation + T-reg suppression → cytokine release (TNF-α, IL-1, IL-6) → endothelial activation + platelet-neutrophil complex formation → ROS/MPO/NETs → diffuse alveolar damage + hyaline membranes.
Phase 2: Proliferative Phase (Days 7–21)
- Type II pneumocytes proliferate to replace destroyed type I cells and attempt barrier repair
- Continued inflammation drives fibroblast recruitment into the alveolar space
- Surfactant dysfunction persists — type II cells produce abnormal surfactant, worsening alveolar instability and collapse
- Organization of the fibrinous exudate begins
Phase 3: Fibrotic Phase (>21 days, in some patients)
- Some patients develop pulmonary fibrosis with obliteration of normal lung architecture
- Ongoing fibrocellular proliferation fills alveoli and interstitium
- Associated with prolonged ventilator dependence and poor prognosis
- Not all patients progress to this phase — resolution can occur after the proliferative phase
Pathophysiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding with protein-rich edema | Impaired O₂ diffusion → hypoxemia |
| Surfactant destruction/dysfunction | Alveolar collapse (atelectasis) → intrapulmonary shunt |
| Microthrombi and vascular obliteration | Increased dead space → hypercapnia |
| Reduced lung compliance | High work of breathing, ventilatory failure |
| Neutrophil-mediated oxidative injury | Further tissue necrosis, delayed repair |
| Non-uniform lung involvement ("baby lung") | Ventilator-induced lung injury (VILI) risk in dependent zones |
Triggers (Direct vs. Indirect)
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma / polytrauma |
| Inhalation injury / toxic gases | Massive blood transfusion (TRALI) |
| Lung contusion | Pancreatitis |
| Near-drowning | Burns |
In direct ARDS, the primary injury targets the alveolar epithelium. In indirect ARDS (e.g., sepsis), systemic inflammatory mediators reach the lung via the bloodstream, injuring the endothelium first — but the downstream cascade converges on the same DAD pattern.
Key Physiological Marker
PaO₂/FiO₂ ratio (P/F ratio) quantifies the severity of gas exchange impairment and drives the Berlin Definition staging:
| Severity | P/F Ratio |
|---|---|
| Mild | 200–300 mmHg |
| Moderate | 100–200 mmHg |
| Severe | <100 mmHg |
In summary, ARDS is a dysregulated innate immune response causing catastrophic breakdown of the alveolar-capillary barrier. The core sequence is: triggering insult → endothelial/epithelial injury → neutrophil-mediated inflammation → alveolar flooding + surfactant failure → severe hypoxemia — culminating in the histological pattern of diffuse alveolar damage.
This is a shared conversation. Sign in to Orris to start your own chat.