Alzheimer disease
Reading File
Finding Sources
Searching PubMed
"Alzheimer disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information to write a thorough response. Let me compile it all.
Alzheimer Disease
Overview
Alzheimer disease (AD) is the most common cause of dementia, accounting for approximately 70% of dementia cases in the United States and 68% of cases in memory disorder clinics. In 1906, Alois Alzheimer first described the hallmark inclusions - amyloid plaques and neurofibrillary tangles (NFTs) - in the brain of a woman in her 50s with paranoia, memory loss, and aphasia. These findings remain the defining pathological features of the disease today.
- Bradley and Daroff's Neurology in Clinical Practice, p. 2049
- Stahl's Essential Psychopharmacology, p. 504
Epidemiology
| Parameter | Data |
|---|---|
| Prevalence (age 65+) | ~11% overall; 3% age 65-74; 32% age 85+ |
| MCI prevalence | Higher in men; AD dementia more prevalent in women |
| Lifetime risk from age 45 | ~10% men, ~20% women |
| Incidence (age 65-74) | 2 new cases per 1,000 per year |
| Incidence (age 75-84) | 11 new cases per 1,000 per year |
| Incidence (age 85+) | 37 new cases per 1,000 per year |
| US projected prevalence by 2060 | ~15 million individuals |
Age is the single most important risk factor. Total payments for all individuals with dementia were estimated at $277 billion in 2018.
- Bradley and Daroff's Neurology in Clinical Practice, p. 2038
Pathology
AD is defined by two cardinal pathological findings:
1. Amyloid Plaques (Extracellular)
Beta-amyloid (Aβ) deposition occurs in a sequential pattern: cortex first, then hippocampus, basal ganglia, thalamus, basal forebrain, and finally brainstem and cerebellum. Plaques are heterogeneous lesions made of extracellular protein deposits. Aβ is derived from amyloid precursor protein (APP) via cleavage by β-secretase and γ-secretase (the presenilin complex).
2. Neurofibrillary Tangles (Intraneuronal)
NFTs are composed of hyperphosphorylated tau protein. Tau normally stabilizes microtubules; when hyperphosphorylated, it aggregates and disrupts neuronal transport. The earliest NFT pathology, according to Braak staging, appears in the locus coeruleus in the third or fourth decade, before reaching limbic regions.
3. Neuronal Loss
Neuronal loss is the third hallmark - so profound it can be seen with the naked eye at autopsy. It predominantly affects limbic and cortical regions, with cholinergic neurons particularly vulnerable. Primary motor, somatosensory, auditory, and visual cortices are relatively spared. The substantia nigra is also relatively spared (distinguishing AD from Parkinson disease).
Histopathology:
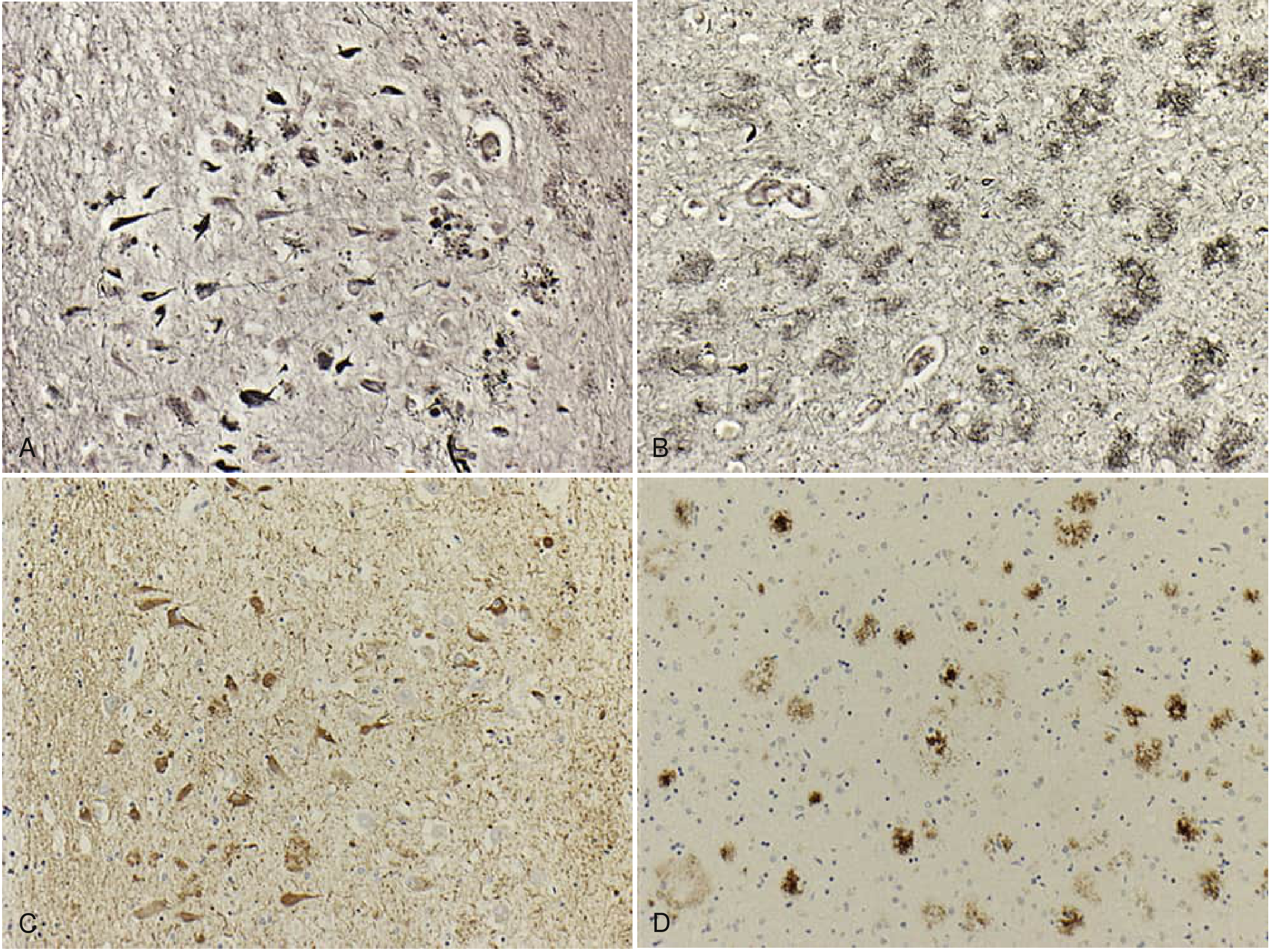
Genetics
The genetic component of AD is estimated at 58-79% (Swedish twin study).
Early-Onset Familial Genes (autosomal dominant, <1% of all AD)
| Gene | Chromosome | Notes |
|---|---|---|
| APP (amyloid precursor protein) | 21 | First described; patients with Down syndrome (trisomy 21) develop AD after age 40 - APP is on chr 21. Mean onset ~age 50. May show myoclonus, seizures, early dyscalculia. |
| PSEN1 (presenilin 1) | 14 | Majority of early-onset familial AD; part of the γ-secretase complex that cleaves APP. May show significant aphasia, myoclonus, seizures. |
| PSEN2 (presenilin 2) | 1 | Rarest autosomal dominant mutation; often in descendants from Volga River region of Russia. Higher seizure rate. |
All three increase brain Aβ levels, supporting the amyloid hypothesis.
Late-Onset Risk Gene
APOE ε4 is the most important genetic risk factor for late-onset AD:
- E4 homozygotes: mean onset age 68, lifetime risk 91%
- E4 heterozygotes: mean onset age 76, lifetime risk 47%
- E4 noncarriers: mean onset age 84, lifetime risk ~20%
- E2 allele: protective against AD
APOE functions in lipid/cholesterol transport from astrocytes to neurons. E4 promotes Aβ aggregation, impairs cerebral Aβ clearance, and causes abnormal lipid transport altering synaptic plasticity.
- Bradley and Daroff's Neurology in Clinical Practice, pp. 27-29
Clinical Features
Typical Amnestic Presentation
AD classically presents with insidious onset progressive memory loss. Cognitive decline begins, on average, 7.5 years before diagnosis. Key features:
- Memory: Rapid forgetting of new information; impaired recognition memory; intrusion errors on recall tasks; absence of normal primacy effect
- Language: Paraphasias, naming deficits (anomia)
- Visuospatial: Impaired construction and spatial perception
- Executive function: Motor planning deficits
- Anosognosia: Impaired awareness of one's own deficits (very common)
DSM-5 Criteria (Major Neurocognitive Disorder due to AD)
- Evidence of significant cognitive decline from previously higher level
- Deficits interfering with everyday functioning
- Insidious onset and gradual progression
- No evidence of other etiology
- Probable AD: objective decline in memory + at least one other cognitive domain
Neuropsychiatric Features
Apathy is the most common neuropsychiatric symptom (72%), followed by depression, agitation, and anxiety.
Atypical Presentations
| Variant | Key Features |
|---|---|
| Posterior Cortical Atrophy (PCA) | Visuospatial/perceptual deficits predominate; occipital-parietal atrophy on MRI |
| Logopenic PPA | Impaired naming, repetition, word retrieval with phonological errors; 64-86% have underlying AD pathology |
| Behavioral/Frontal variant AD | Early executive dysfunction, apathy (vs. hyperorality/perseverative behaviors in bvFTD) |
Diagnostic Criteria and Biomarkers
The AT(N) Research Framework (2018 NIA-AA) classifies individuals biologically by three biomarker groups:
| Biomarker | Positive Finding | Measured by |
|---|---|---|
| A - Amyloid deposition | Aβ42↓ in CSF or amyloid PET+ | CSF or PET |
| T - Pathological tau | Phospho-tau↑ in CSF or tau PET+ | CSF or tau PET |
| (N) - Neurodegeneration | FDG-PET hypometabolism, MRI atrophy | PET or MRI |
- Preclinical AD = A+T+, cognitively normal
- Prodromal AD = A+T+, MCI stage
- AD dementia = A+T+, dementia stage
Neuroimaging
FDG-PET shows progressive posterior glucose hypometabolism (temporo-parietal regions):
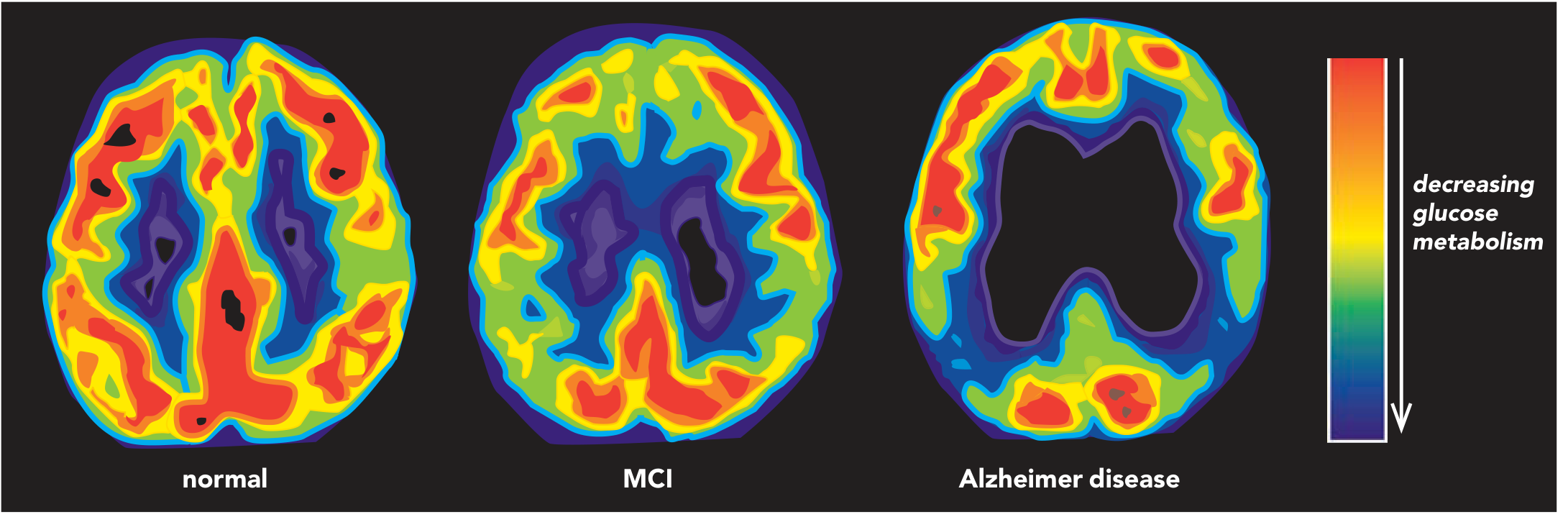
MRI in mild AD shows:
- 20-30% loss of entorhinal cortex volume
- 15-25% loss of hippocampal volume
- Ventricular enlargement
Updated 2025 appropriate use criteria for amyloid and tau PET have been published by the Alzheimer's Association and SNMMI (PMID: 39776249).
Treatment
1. Cholinesterase Inhibitors (AChE-I)
Indicated for mild to moderate AD. Based on the cholinergic deficit hypothesis - AD causes profound loss of cholinergic neurons in the nucleus basalis of Meynert.
| Drug | Mechanism | Starting Dose | Target Dose |
|---|---|---|---|
| Donepezil | Selective, reversible AChE-I | 5 mg daily | 10 mg daily |
| Rivastigmine | AChE-I + BuChE-I | 1.5 mg bd (oral) or 4.6 mg/24h patch | 3-6 mg bd or 9.5 mg/24h patch |
| Galantamine | Competitive AChE-I + nicotinic receptor agonist | 8 mg XL daily | 16-24 mg XL daily |
Common adverse effects: nausea, vomiting, diarrhoea, anorexia, insomnia, muscle cramps, bradycardia.
2. Memantine
Indicated for moderate to severe AD. Acts as a low-to-moderate affinity, non-competitive NMDA receptor antagonist - binds preferentially to open NMDA receptor-operated calcium channels, blocking pathologically elevated glutamate-mediated excitotoxicity.
- Starting dose: 5 mg daily; titrate weekly by 5 mg
- Target dose: 20 mg daily (or 10 mg twice daily)
- Can be combined with AChE-I in moderate-severe disease
3. Disease-Modifying Therapy (Anti-Amyloid Monoclonal Antibodies)
A major paradigm shift has occurred with FDA-approved anti-amyloid immunotherapy:
- Lecanemab (Leqembi): Approved 2023 - anti-Aβ protofibrils; shown to reduce amyloid burden and slow cognitive decline in early AD
- Donanemab: Approved 2024 - targets a modified form of Aβ (N3pG)
These agents are associated with ARIA (amyloid-related imaging abnormalities) - edema (ARIA-E) and microhemorrhages (ARIA-H) - which require MRI monitoring.
4. Non-Pharmacological & Preventive Approaches
A 2025 meta-analysis (PMID: 39797935) confirmed the Mediterranean diet significantly reduces risk of cognitive impairment and AD. Vascular risk factor control (hypertension, diabetes, dyslipidemia), physical exercise, cognitive engagement, and social activity remain important modifiable factors.
Stages of AD
| Stage | Clinical Features |
|---|---|
| Preclinical AD | Biomarker positive (A+ and/or T+), cognitively normal |
| MCI due to AD | Memory impairment + normal ADLs; biomarker evidence of AD |
| Mild AD dementia | Cognitive impairment affecting some daily activities |
| Moderate AD dementia | Greater memory and functional decline; behavioral symptoms emerge |
| Severe AD dementia | Profound cognitive loss; incontinence; loss of ambulation; death typically from aspiration pneumonia or infection |
Key Pathophysiological Hypotheses
-
Amyloid cascade hypothesis: Overproduction or impaired clearance of Aβ42 leads to oligomers and plaques, triggering tau hyperphosphorylation, NFT formation, synaptic dysfunction, and neuronal death. All three familial gene mutations (APP, PSEN1, PSEN2) increase Aβ, supporting this hypothesis.
-
Tau hypothesis: Hyperphosphorylated tau spreads trans-synaptically in a prion-like manner to anatomically connected regions, driving neurodegeneration independently.
-
Cholinergic hypothesis: Loss of cholinergic neurons in the basal forebrain (nucleus basalis of Meynert) causes memory and cognitive deficits - the basis for AChE-I therapy.
-
Glutamate excitotoxicity: Sustained excessive NMDA receptor activation causes neuronal dysfunction and death - the rationale for memantine.
Recent Evidence Update (2025):
- Updated appropriate use criteria for amyloid and tau PET imaging have been published (PMID: 39776249), guiding clinical use of these biomarker tools.
- Clinical criteria for LATE (limbic-predominant age-related TDP-43 encephalopathy) have been developed (PMID: 39807681) - an important differential diagnosis/comorbidity that can mimic or co-occur with AD.
- Mediterranean diet adherence is associated with significant reduction in AD risk (PMID: 39797935).
Sources: Bradley and Daroff's Neurology in Clinical Practice; Stahl's Essential Psychopharmacology; The Maudsley Prescribing Guidelines in Psychiatry (15th ed.); Thompson & Thompson Genetics and Genomics in Medicine.
This is a shared conversation. Sign in to Orris to start your own chat.