enlist each and every inborn error of protein metablism along with pathways,clinical findings,defective enzymes,treatment,symptoms as well as images of symptoms
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Searching the Web
inborn errors protein metabolism clinical photos symptoms PKU alkaptonuria MSUD albinism homocystinuria
Reading File
Loading Image
Loading Image
Loading Image
Searching the Web
alkaptonuria ochronosis ear cartilage clinical photo homocystinuria lens dislocation MSUD clinical image
Reading File
Loading Image
Loading Image
Now I have comprehensive data from the textbooks. Let me compile the full, detailed answer.
Inborn Errors of Protein (Amino Acid) Metabolism
These are inherited (usually autosomal recessive) disorders caused by enzyme defects in amino acid catabolic pathways. They collectively affect ~1 in 4,000 newborns. The disease name reflects what accumulates: parent amino acid in aminoacidopathies (-emias/-urias), or downstream metabolites in organic acidemias.
Overview Table (Harrison's 22E, Table 431-1)
| Amino Acid | Condition | Defective Enzyme | Key Clinical Findings | Inheritance |
|---|---|---|---|---|
| Phenylalanine | Phenylketonuria (PKU) | Phenylalanine hydroxylase | Intellectual disability, eczema, "mousy" odor, hypopigmentation | AR |
| Phenylalanine | DHPR deficiency | Dihydropteridine reductase | Intellectual disability, microcephaly | AR |
| Phenylalanine | PTPS deficiency | 6-Pyruvoyltetrahydropterin synthase | Dystonia, neurologic deterioration, seizures | AR |
| Tyrosine | Tyrosinemia type I | Fumarylacetoacetate hydrolase | Liver failure, cirrhosis, renal tubular acidosis, peripheral neuropathy | AR |
| Tyrosine | Tyrosinemia type II | Tyrosine aminotransferase | Palmoplantar keratoderma, corneal erosions, photophobia | AR |
| Tyrosine | Tyrosinemia type III | 4-Hydroxyphenylpyruvate dioxygenase | Hypertriglyceridemia, occasional mental delay | AR |
| Tyrosine | Alkaptonuria | Homogentisate oxidase | Dark urine, ochronosis, arthritis, cardiac valve involvement | AR |
| Tyrosine | Albinism (OCA) | Tyrosinase | Hypopigmentation of hair/skin/eyes, photophobia, ↑ skin cancer risk | AR |
| Methionine/Homocysteine | Homocystinuria (classical) | Cystathionine β-synthase | Ectopia lentis, Marfanoid habitus, intellectual disability, thrombosis | AR |
| Methionine/Homocysteine | Remethylation defects | Methionine synthase/MTHFR | Developmental delay, megaloblastic anemia, low methionine | AR |
| Branched-chain AA | MSUD | Branched-chain α-ketoacid dehydrogenase (BCKD) | Maple syrup urine odor, encephalopathy, poor feeding | AR |
| Urea cycle | OTC deficiency | Ornithine transcarbamylase | Hyperammonemia, coma, protein aversion | X-linked |
| Urea cycle | CPS-1 deficiency | Carbamoyl phosphate synthase-1 | Hyperammonemia, lethargy, coma | AR |
| Urea cycle | Citrullinemia | Argininosuccinate synthase | Hyperammonemia, very high citrulline | AR |
| Urea cycle | Argininosuccinic aciduria | Argininosuccinate lyase | Hyperammonemia, trichorrhexis nodosa (brittle hair) | AR |
| Urea cycle | Arginase deficiency | Arginase | Spastic diplegia, intellectual disability, high arginine | AR |
1. Phenylketonuria (PKU)
Pathway: Phenylalanine → Tyrosine (blocked) → accumulation of phenylalanine → overflow to phenylpyruvate, phenyllactate, phenylacetate
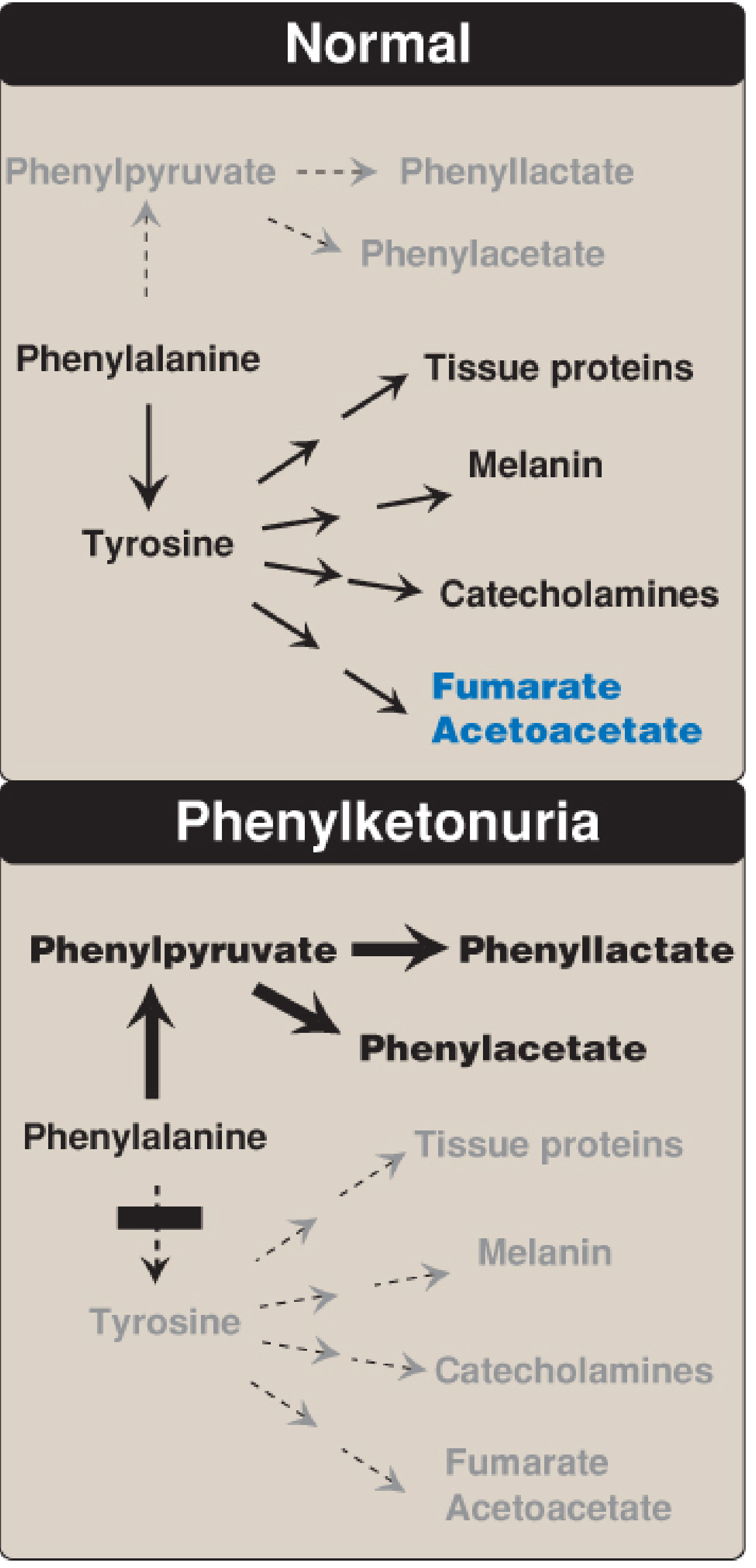
Defective Enzyme: Phenylalanine hydroxylase (PAH) - requires tetrahydrobiopterin (BH4) as cofactor
Accumulates: Phenylalanine, phenylpyruvate, phenyllactate, phenylacetate
Symptoms/Clinical Findings:
- Intellectual disability (if untreated) - phenylalanine is toxic to the developing brain
- Microcephaly
- Seizures (in untreated cases)
- Hypopigmentation - fair hair, pale skin, blue eyes (reduced tyrosine → reduced melanin)
- Eczema-like skin rash
- "Mousy" or musty odor of urine/sweat (phenylacetate)
- Behavioral disturbances

Treatment:
- Low-phenylalanine diet (started within first 7-10 days of life)
- Tyrosine supplementation (becomes conditionally essential)
- Oral tetrahydrobiopterin (BH4/sapropterin) 5-20 mg/kg/day - reduces phenylalanine levels in BH4-responsive patients
- Enzyme substitution with phenylalanine ammonia lyase (pegvaliase) for adults
Diagnosis: Newborn screening (Guthrie test / tandem MS) - blood phenylalanine >120 μmol/L
Inheritance: Autosomal recessive; >100 different mutations in PAH gene known
2. BH4 Deficiency Variants (Hyperphenylalaninemias)
These mimic PKU but affect BH4 synthesis/recycling, causing more severe neurological damage since BH4 is also required for tyrosine hydroxylase and tryptophan hydroxylase (neurotransmitter synthesis).
| Variant | Defective Enzyme | Extra Features |
|---|---|---|
| DHPR deficiency | Dihydropteridine reductase | Microcephaly, neurological deterioration even on low-Phe diet |
| PTPS deficiency | 6-Pyruvoyltetrahydropterin synthase | Dystonia, oculomotor problems |
| GTP-CH1 deficiency | GTP cyclohydrolase 1 | Temperamental instability, dentation defects |
| PCD deficiency | Pterin-4α-carbinolamine dehydratase | Transient benign hyperphenylalaninemia |
Treatment: BH4 supplementation + neurotransmitter precursors (L-DOPA + 5-hydroxytryptophan)
3. Tyrosinemias
Type I (Hepatorenal Tyrosinemia / Tyrosinosis)
Pathway: Tyrosine → 4-hydroxyphenylpyruvate → homogentisate → maleylacetoacetate → fumarylacetoacetate → (BLOCKED) → accumulation of succinylacetone
Defective Enzyme: Fumarylacetoacetate hydrolase (FAH)
Symptoms/Clinical Findings:
- Liver failure and cirrhosis (most prominent)
- Hepatocellular carcinoma risk
- Renal tubular Fanconi syndrome (renal tubular acidosis, glycosuria, phosphaturia)
- Peripheral neuropathy (painful crises - "cabbage" odor)
- Elevated AFP (useful marker)
Treatment: Nitisinone (NTBC) blocks 4-hydroxyphenylpyruvate dioxygenase (upstream), preventing toxic metabolite accumulation. Diet low in phenylalanine and tyrosine. Liver transplantation in severe cases.
Type II (Oculocutaneous Tyrosinemia / Richner-Hanhart Syndrome)
Defective Enzyme: Tyrosine aminotransferase (TAT)
Symptoms:
- Painful palmoplantar hyperkeratosis
- Corneal erosions and dendritic ulcers, photophobia
- Mild intellectual disability in some
Treatment: Low-tyrosine, low-phenylalanine diet
Type III
Defective Enzyme: 4-Hydroxyphenylpyruvate dioxygenase (HPPD)
Symptoms: Mild neurological problems, hypertriglyceridemia; generally mild
4. Alkaptonuria
Pathway: Tyrosine → 4-hydroxyphenylpyruvate → homogentisate → (BLOCKED) → homogentisate accumulates and polymerizes in connective tissues
Defective Enzyme: Homogentisate 1,2-dioxygenase (homogentisic acid oxidase)

Classic Triad of Symptoms:
- Homogentisic aciduria - urine turns dark/black on standing (oxidation of homogentisic acid); earliest sign is dark-stained diapers in infants
- Ochronosis - blue-black pigmentation of cartilage (ear cartilage, intervertebral discs), sclera, and skin
- Arthritis - progressive large-joint and spinal osteoarthritis
Other Features:
- Cardiac valve involvement (mitral/aortic stenosis)
- Coronary artery calcification
- Urinary/prostatic stones
Treatment:
- No curative treatment
- Dietary restriction of phenylalanine and tyrosine (slows accumulation)
- Nitisinone (experimental - blocks upstream HPPD, reduces HGA production)
- Symptomatic management: NSAIDs, joint replacement
- Ascorbic acid (vitamin C) - may slow ochronosis
Inheritance: Autosomal recessive; incidence ~1:250,000
5. Albinism
Pathway: Tyrosine → (tyrosinase, step 1) → DOPA → (tyrosinase, step 2) → DOPAquinone → Melanin (BLOCKED at step 1 or 2)
Defective Enzyme (OCA Type 1): Tyrosinase (copper-containing enzyme)
Types:
| Type | Gene | Feature |
|---|---|---|
| OCA1 (Tyrosinase-negative) | TYR | Complete absence of melanin; most severe |
| OCA2 | OCA2/P gene | Variable pigmentation; most common worldwide |
| OCA3 | TYRP1 | Brown OCA; reddish-brown skin |
| OCA4 | SLC45A2 | Variable; common in Japan |
| Ocular albinism | GPRA/other | Eyes only affected; X-linked form exists |
Symptoms:
- Generalized hypopigmentation: white/pale hair, pink/white skin
- Red pupils (light reflected from retinal vessels due to absent iris pigment)
- Nystagmus, photophobia
- Reduced visual acuity
- Misrouting of optic nerve fibers
- Markedly increased risk of skin cancer (UV damage)
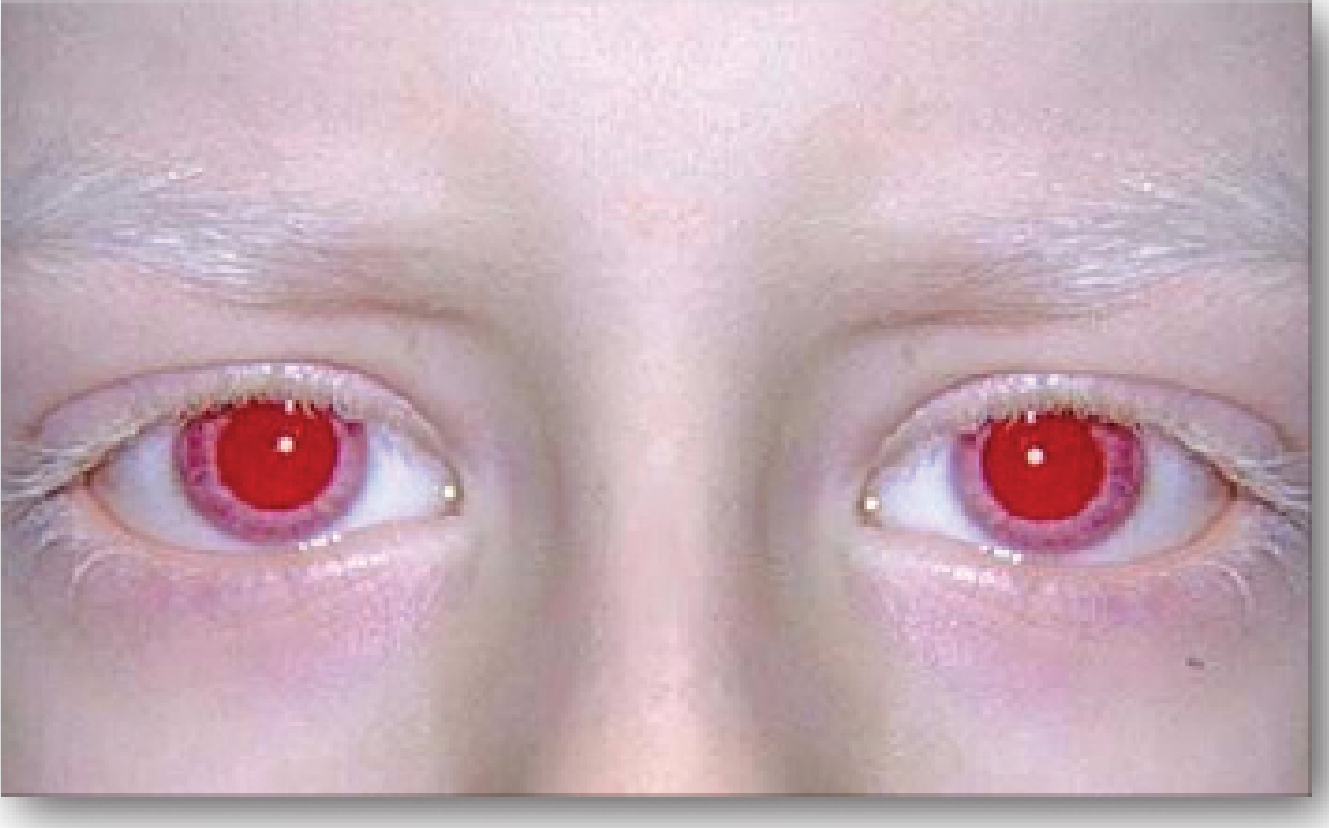
Treatment:
- Sun protection (SPF 50+ sunscreen, protective clothing)
- Corrective lenses and low-vision aids
- No biochemical cure; gene therapy under investigation
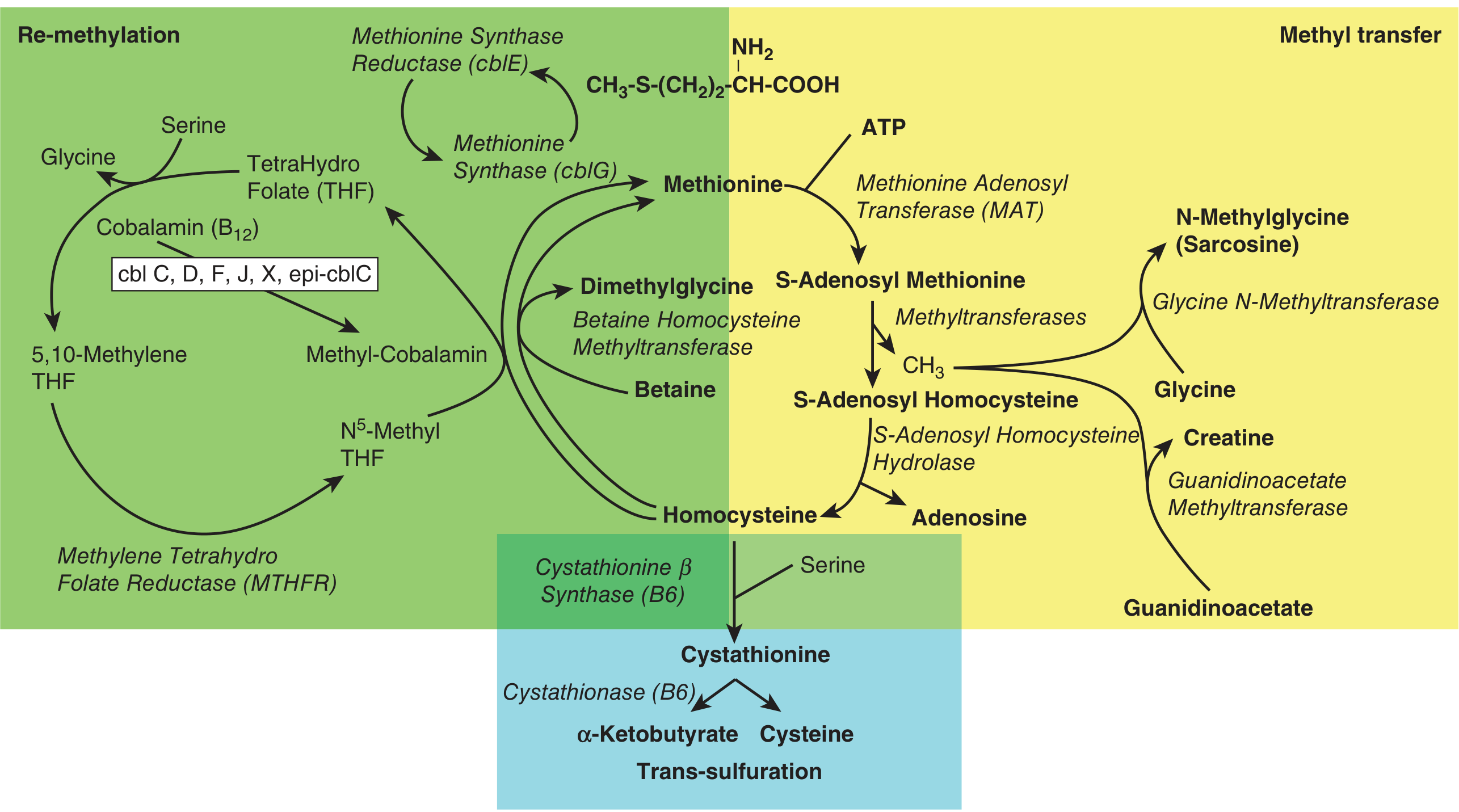
6. Homocystinuria
Pathway: Methionine → SAM → SAH → Homocysteine → (BLOCKED at cystathionine β-synthase) → homocysteine accumulates
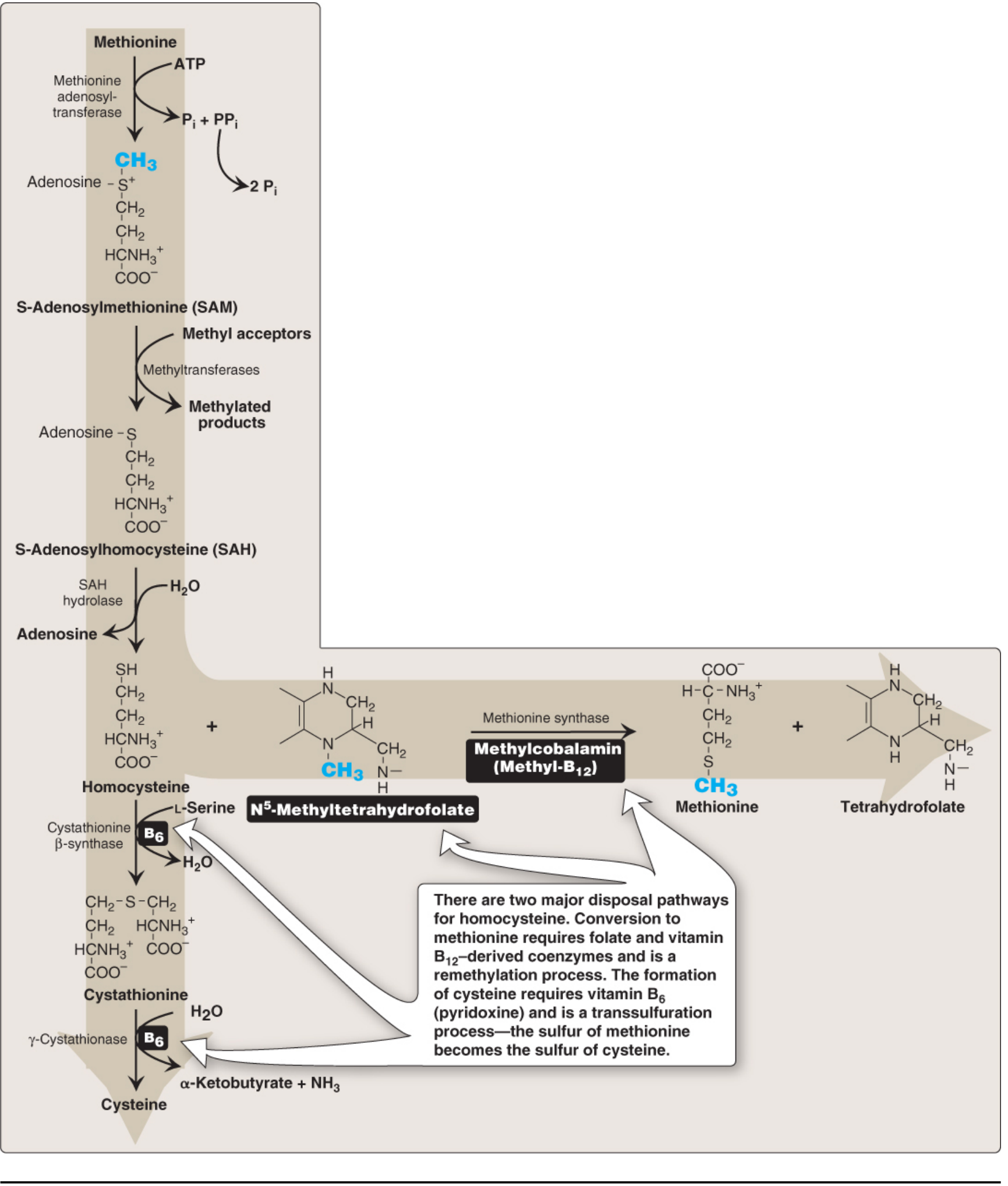
Classic (Type I) - Cystathionine β-Synthase Deficiency:
Defective Enzyme: Cystathionine β-synthase (CBS) - pyridoxal phosphate (B6)-dependent; condenses homocysteine + serine → cystathionine
Symptoms/Clinical Findings (present at 3-5 years):
- Ectopia lentis (downward dislocation of lens) - distinguishes from Marfan syndrome where lens displaces upward
- Marfanoid habitus - tall, long limbs, arachnodactyly
- Intellectual disability (~50% of cases)
- Osteoporosis
- Thromboembolic events (major cause of morbidity/mortality) - affects coronary, renal, and cerebral vessels even in children
- Elevated plasma methionine and free homocysteine (total plasma Hcy usually >100 μmol/L)
Treatment:
- Pyridoxine (B6) 25-500 mg/day - ~50% of patients are B6-responsive (milder phenotype)
- Low-methionine diet
- Folate and vitamin B12 supplementation
- Betaine (trimethylglycine) - promotes remethylation of Hcy → methionine
- Cysteine supplementation (becomes essential amino acid)
Homocystinuria (Remethylation Defects - Types II-X):
| Subtype | Defect | Key Feature |
|---|---|---|
| MTHFR deficiency | Methylenetetrahydrofolate reductase | Low methionine, developmental delay |
| Methionine synthase (cblG) deficiency | Methionine synthase | Megaloblastic anemia |
| Methionine synthase reductase (cblE) deficiency | Methionine synthase reductase | Megaloblastic anemia |
| cblC, cblD, cblF, cblJ | Cobalamin processing enzymes | Combined methylmalonic acidemia + homocystinuria |
Treatment of remethylation defects: Methylfolate + hydroxycobalamin (activated B12) + betaine
7. Maple Syrup Urine Disease (MSUD)
Pathway: Leucine, Isoleucine, Valine → (transamination) → α-keto acids → (BLOCKED at oxidative decarboxylation) → branched-chain keto acids and amino acids accumulate
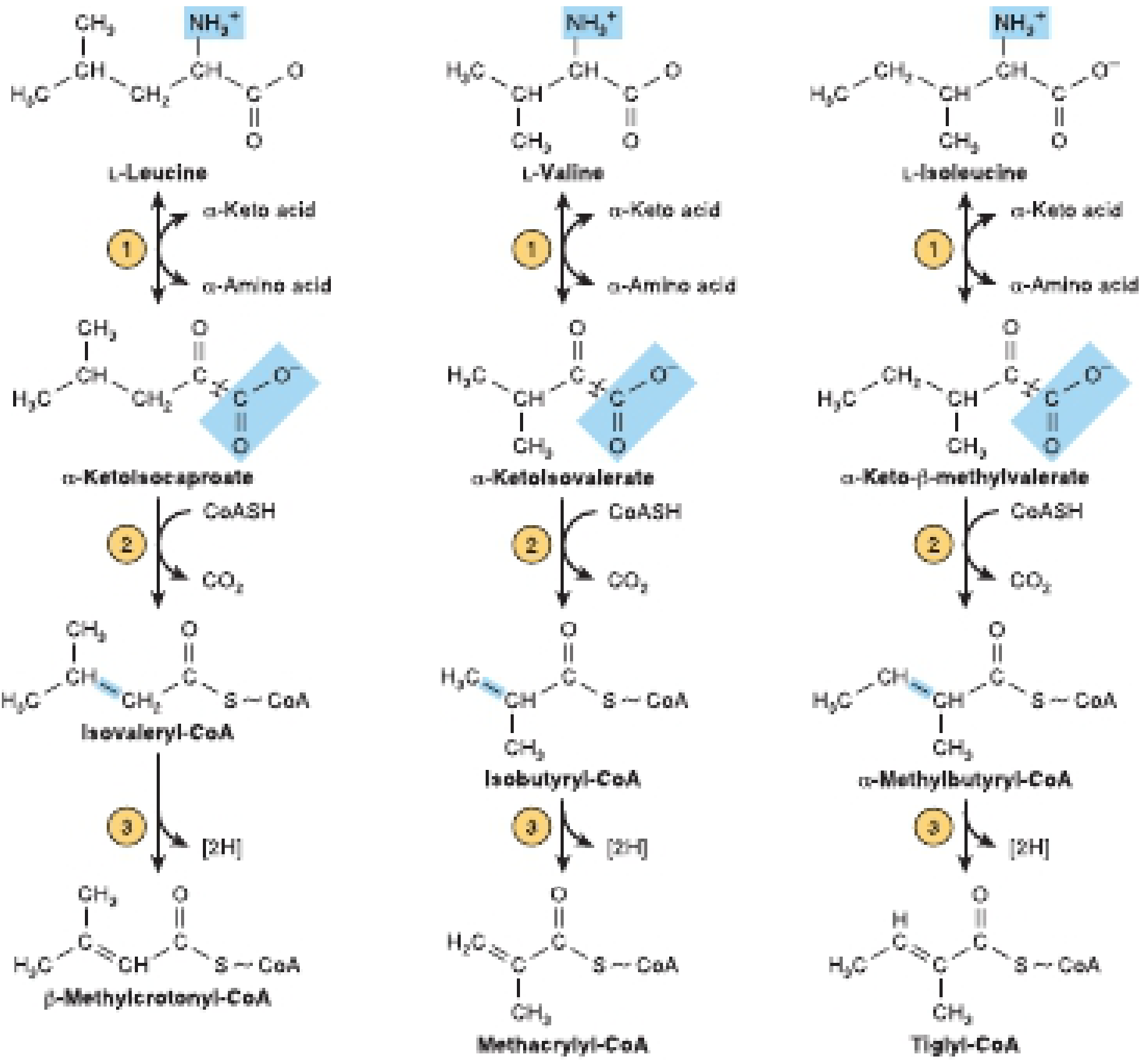
Defective Enzyme: Branched-chain α-ketoacid dehydrogenase (BCKD) complex - a multi-enzyme complex requiring thiamine (B1), lipoic acid, CoA
BCKD has 4 subunits (E1α, E1β, E2, E3); defects in any cause MSUD:
| Component | Type |
|---|---|
| E1α (BCKDHA) | Classic MSUD |
| E1β (BCKDHB) | Intermediate MSUD |
| E2 (DBT) | Intermediate MSUD |
| E3 (DLD) | E3-deficient MSUD (also affects pyruvate dehydrogenase and α-ketoglutarate dehydrogenase) |
Accumulates: Leucine, isoleucine, valine (BCAAs), alloisoleucine (pathognomonic), and their α-keto acid derivatives
Symptoms/Clinical Findings:
- Maple syrup / burnt sugar odor of urine, cerumen, and sweat (due to sotolone in keto acids)
- Neonatal encephalopathy (2-5 days of life): poor feeding, vomiting, lethargy
- Hypotonia progressing to hypertonia, opisthotonus
- Seizures
- Leucine toxicity is the primary neurotoxic mechanism
- Cerebral edema on MRI (T2 signal abnormalities in basal ganglia, brainstem, cerebellum)
- Without treatment: death or severe intellectual disability
Treatment:
- Synthetic BCAA-free formula with monitored small amounts of leucine, isoleucine, valine
- Thiamine (B1) supplementation (some patients are thiamine-responsive at 10-1000 mg/day)
- Acute decompensation: IV glucose (suppress catabolism), hemodialysis (rapidly lower leucine)
- Liver transplantation - effectively cures the metabolic defect (provides sufficient BCKD enzyme)
- Lifelong dietary management; leucine levels carefully monitored
8. Urea Cycle Defects
Pathway: NH3 → Carbamoyl phosphate → Citrulline → Argininosuccinate → Arginine → Urea (+ Ornithine recycled)
Each step has a corresponding inborn error:
| Step | Enzyme | Disease | Key Feature |
|---|---|---|---|
| Step 1 | N-Acetylglutamate synthase (NAGS) | NAGS deficiency | Severe neonatal hyperammonemia |
| Step 2 | Carbamoyl phosphate synthase 1 (CPS-1) | CPS-1 deficiency | Severe hyperammonemia; low citrulline, low orotic acid |
| Step 3 | Ornithine transcarbamylase (OTC) | OTC deficiency (most common) | X-linked; boys severely affected; ↑ orotic acid in urine |
| Step 4 | Argininosuccinate synthase (ASS) | Citrullinemia type I | Markedly elevated citrulline |
| Step 5 | Argininosuccinate lyase (ASL) | Argininosuccinic aciduria | High argininosuccinic acid; trichorrhexis nodosa |
| Step 6 | Arginase-1 | Arginase deficiency | Spastic diplegia, hyperargininemia (hyperammonemia less severe) |
| Transport | Ornithine transporter ORNT1 | HHH syndrome | Hyperammonemia-hyperornithinemia-homocitrullinuria |
| Transport | Citrin (SLC25A13) | Citrullinemia type II | Adult-onset; liver disease |
Symptoms (all urea cycle defects share):
- Hyperammonemia - the core toxicity
- Neonatal presentation (complete defects): refusal to eat, lethargy → coma → death at 1-4 days
- Partial defects: protein aversion, recurrent vomiting, migraine, behavioral changes, disorientation
- Elevated plasma glutamine (ammonia detox via perivenous hepatocytes)
- Chronic liver dysfunction
Diagnosis: Plasma ammonia + plasma amino acids + urine orotic acid profile (orotic acid distinguishes OTC deficiency from CPS-1/NAGS)
Treatment:
- Protein restriction (0.5-1 g/kg/day)
- Nitrogen scavengers: Sodium benzoate + sodium phenylacetate (Ammonul) - provide alternative pathways for nitrogen excretion
- Arginine and/or citrulline supplementation (become essential; also stimulate cycle flux)
- Acute hyperammonemia: IV glucose + lipids, hemodialysis for ammonia >500 μmol/L
- Liver transplantation - definitive cure for most urea cycle defects
- Gene therapy under investigation
9. Other Notable Inborn Errors
Cystinuria (Cystine Transport Defect)
- Defect: SLC3A1 / SLC7A9 genes - defective transporter for cystine, ornithine, arginine, lysine in renal tubules and gut
- Symptoms: Recurrent urinary cystine stones (radiopaque), renal colic, obstructive uropathy
- Treatment: High fluid intake, urine alkalinization, D-penicillamine or tiopronin (chelate cystine)
Hartnup Disease (Tryptophan Transport Defect)
- Defect: SLC6A19 - neutral amino acid transporter; impaired intestinal/renal tryptophan absorption
- Pathway: Tryptophan → nicotinamide (NAD precursor) pathway impaired
- Symptoms: Pellagra-like rash (photosensitive), cerebellar ataxia, psychiatric symptoms; often intermittent
- Treatment: High-protein diet, niacinamide supplementation
Isovaleric Acidemia
- Defect: Isovaleryl-CoA dehydrogenase (leucine catabolism)
- Symptoms: "Sweaty feet" odor (isovalerate), metabolic acidosis, hyperammonemia, bone marrow suppression, encephalopathy
- Treatment: Low-leucine diet, glycine supplementation (conjugates isovaleryl-CoA), carnitine
Propionic Acidemia
- Defect: Propionyl-CoA carboxylase (biotin-dependent) - valine/isoleucine/methionine/threonine catabolism
- Symptoms: Neonatal ketoacidosis, hyperammonemia, hypotonia, cardiomyopathy
- Treatment: Low-protein diet, biotin, carnitine, liver transplantation
Methylmalonic Acidemia (MMA)
- Defect: Methylmalonyl-CoA mutase (requires adenosylcobalamin/B12) OR cobalamin metabolism enzymes
- Symptoms: Neonatal ketoacidosis, hyperammonemia, renal failure (long-term), optic atrophy, metabolic stroke
- Treatment: Low-protein diet, vitamin B12 (cobalamin-responsive forms), carnitine; liver/kidney transplantation
Nonketotic Hyperglycinemia (NKH)
- Defect: Glycine cleavage system (P-protein / H-protein / T-protein / L-protein)
- Symptoms: Neonatal seizures, profound hypotonia, burst-suppression EEG, hiccups, apnea, intellectual disability
- Treatment: Sodium benzoate (scavenges glycine), dextromethorphan (NMDA antagonist); poor prognosis
Hyperprolinemia
- Type I: Proline oxidase deficiency - generally benign
- Type II: Δ1-Pyrroline-5-carboxylate dehydrogenase deficiency - seizures, intellectual disability
- Symptoms: Elevated plasma/urine proline
Hydroxyprolinemia
- Defect: 4-Hydroxyproline dehydrogenase - benign, no clinical consequences
Sarcosinemia
- Defect: Sarcosine dehydrogenase - no clinical consequences; benign biochemical finding
Key Diagnostic Concepts
| Marker | Disorder |
|---|---|
| ↑ Phenylalanine on newborn screen | PKU and BH4 defects |
| Dark/black urine on standing | Alkaptonuria |
| Maple syrup odor + alloisoleucine | MSUD |
| ↑ NH3 + ↑ orotic acid | OTC deficiency |
| ↑ NH3 + low/no orotic acid | CPS-1 or NAGS deficiency |
| ↑ Homocysteine + ↑ methionine | CBS deficiency (homocystinuria) |
| ↑ Homocysteine + ↓ methionine | Remethylation defects |
| ↑ Succinylacetone in urine | Tyrosinemia type I |
| Cystine crystals in urine | Cystinuria |
Summary Pathway Diagram
The major phenylalanine/tyrosine disease cluster in one pathway:
Phenylalanine
↓ [PAH + BH4] ← PKU (PAH defect)
Tyrosine
↓ [TAT] ← Tyrosinemia II (TAT defect)
4-OH-phenylpyruvate
↓ [HPPD] ← Tyrosinemia III (HPPD defect)
Homogentisate
↓ [HGD] ← Alkaptonuria (HGD defect)
Maleylacetoacetate
↓
Fumarylacetoacetate
↓ [FAH] ← Tyrosinemia I (FAH defect)
Fumarate + Acetoacetate
Tyrosine → [Tyrosinase] → DOPA → Melanin ← Albinism (Tyrosinase defect)
Tyrosine → [TH + BH4] → DOPA → Catecholamines
Sources: Harrison's Principles of Internal Medicine 22E (2025); Biochemistry 8th Ed Lippincott Illustrated Reviews; Harper's Illustrated Biochemistry 32nd Ed; Basic Medical Biochemistry 6th Ed; Thompson & Thompson Genetics and Genomics 9th Ed; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.