multiple myeloma
Reading File
Finding Sources
Searching PubMed
"multiple myeloma"[MeSH Terms] AND "therapy"[MeSH Terms]
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Here is a comprehensive overview of Multiple Myeloma, drawn from Goldman-Cecil Medicine, Robbins & Kumar Pathologic Basis of Disease, and Campbell's Operative Orthopaedics.
Multiple Myeloma
Definition and Epidemiology
Multiple myeloma is a malignant neoplasm of clonal plasma cells that proliferate in the bone marrow and produce a monoclonal immunoglobulin (M protein). It is one of the most common lymphoid malignancies, with approximately 30,000 new cases per year in the United States. The median age at diagnosis is 70 years. It is more common in males and occurs at higher frequency in people of African descent. - Robbins & Kumar Basic Pathology
Pathogenesis
Myeloma frequently arises from chromosomal translocations fusing the IgH locus on chromosome 14 to oncogenes such as cyclin D1 and cyclin D3, leading to dysregulated cell proliferation. Key molecular drivers include:
- IL-6 (produced by bone marrow stromal cells) promotes plasma cell proliferation
- RANKL upregulation by myeloma-derived factors activates osteoclasts, driving bone resorption while simultaneously inhibiting osteoblast function
- Late-stage disease may show MYC translocations, associated with aggressive behavior
- High-risk cytogenetics: deletions of 13q, 17p, and the t(4;14) translocation portend a worse prognosis
- Favorable: translocations involving cyclin D1
Clinical Features (CRAB Criteria)
The classic mnemonic CRAB captures the major end-organ effects:
| Feature | Mechanism |
|---|---|
| C - Hypercalcemia (15-20% at diagnosis) | Osteoclast activation, bone resorption |
| R - Renal failure | Light chain cast nephropathy, amyloid deposition, hypercalcemia |
| A - Anemia | Marrow replacement by plasma cells |
| B - Bone lesions | Punched-out lytic lesions, pathologic fractures |
Additional features include:
- Bone pain (most common complaint) - spine, ribs, pelvis most affected
- Recurrent bacterial infections (due to suppression of normal immunoglobulin production despite high total Ig levels)
- Hyperviscosity syndrome (~7% of patients, especially IgA or IgG3 subtypes)
- Peripheral neuropathy (especially with osteosclerotic myeloma)
Immunoglobulin Profile
- IgG: ~55-60% (most common)
- IgA: ~20-25%
- Light chains only (Bence Jones proteins): ~20%
- IgM, IgD, IgE: rare
- Nonsecretory myeloma: ~1% - absence of M protein does not exclude diagnosis
Diagnosis
Diagnosis requires both:
- Clonal plasma cells in bone marrow (>10%, or >30% by some criteria)
- End-organ damage (CRAB criteria) OR myeloma-defining biomarkers
Key investigations:
- Serum protein electrophoresis (SPEP): shows a sharp monoclonal "spike" (M protein >3 g/dL typical)
- Immunofixation: characterizes the heavy and light chain class (e.g., IgGκ)
- Urine protein electrophoresis: detects Bence Jones protein (>6 mg/dL typical)
- Serum free light chain (FLC) ratio: included in 2014 IMWG diagnostic criteria - involved FLC >100 mg/L is myeloma-defining
- Bone marrow biopsy: required for definitive diagnosis
- Skeletal survey / PET-CT / MRI: staging and detecting lytic lesions
- CBC, serum creatinine, calcium, LDH, beta-2 microglobulin: staging markers
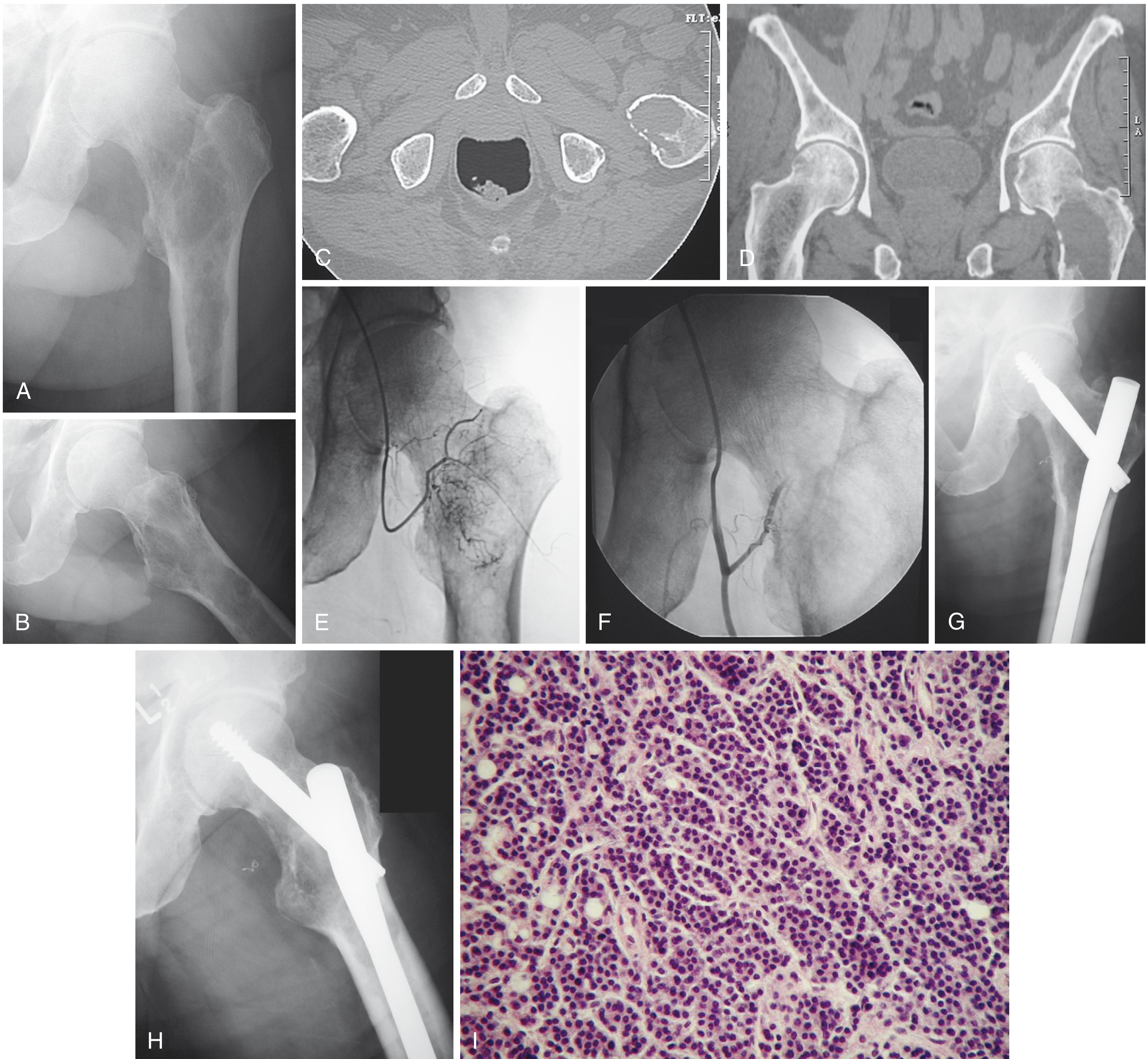
Morphology
On bone marrow biopsy, myeloma cells are sheets of plasma cells - small round blue cells with:
- "Clock-face" (cartwheel) chromatin pattern
- Abundant basophilic cytoplasm with perinuclear "halo"
- Prominent nucleoli and Russell bodies (cytoplasmic Ig inclusions) in some cases
- Plasma cells >30% of marrow cellularity
Radiographically, lesions appear as multiple punched-out lytic defects (1-4 cm), without surrounding reactive sclerosis. Most lesions are negative on bone scan (due to absent osteoblast activity).
Staging
The Revised International Staging System (R-ISS) incorporates:
- Serum beta-2 microglobulin
- Serum albumin
- LDH
- High-risk cytogenetics (del 17p, t(4;14), t(14;16))
Treatment
Treatment is stratified by transplant eligibility:
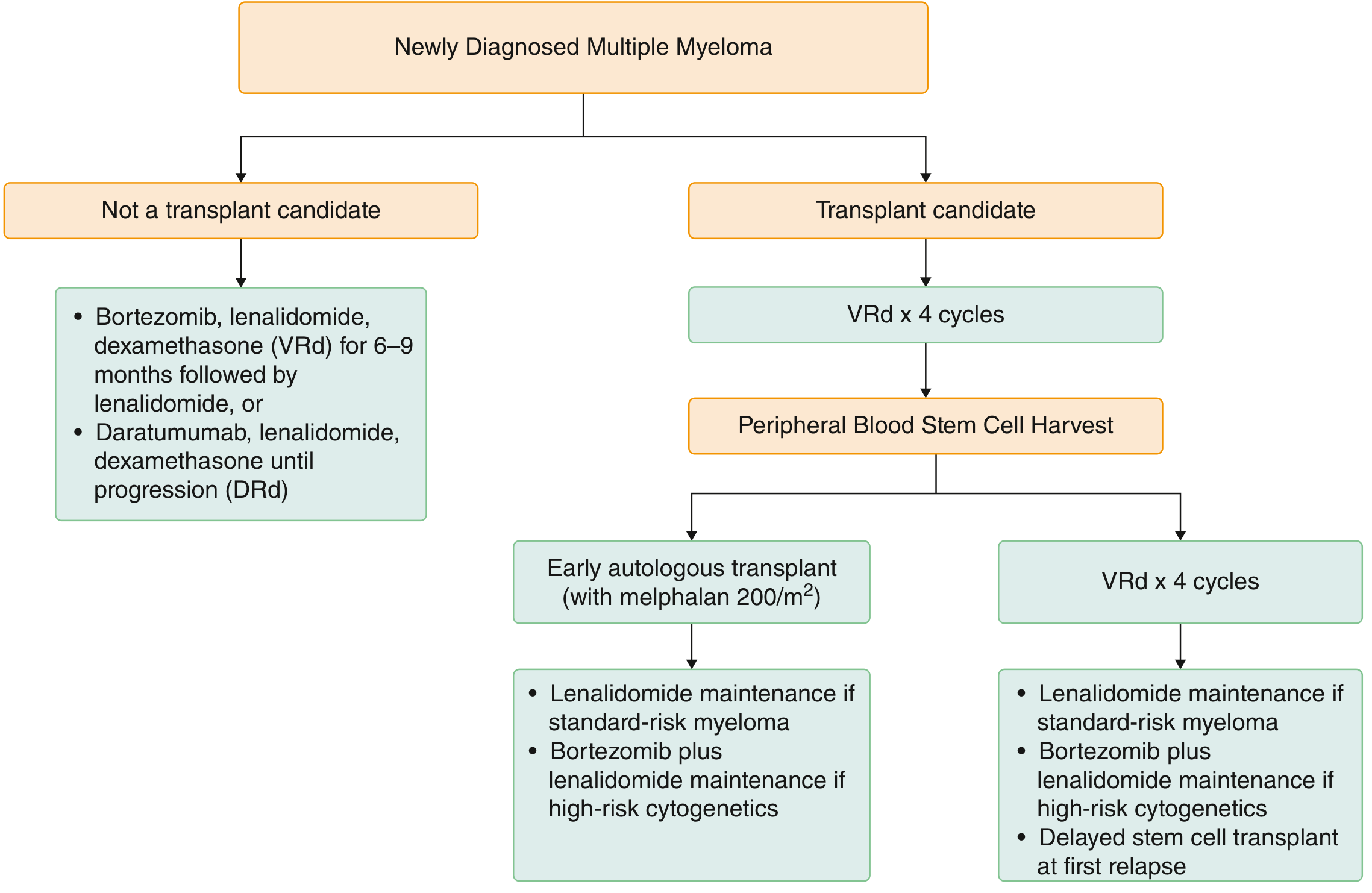
Transplant-Eligible Patients (~50%)
- Induction: VRd (bortezomib + lenalidomide + dexamethasone) x 3-4 cycles; daratumumab can be added for high-risk disease
- Stem cell harvest with G-CSF ± plerixafor
- Conditioning: Melphalan 200 mg/m² followed by autologous stem cell infusion
- Maintenance: Lenalidomide (standard-risk); bortezomib + lenalidomide (high-risk cytogenetics)
- Early vs. delayed transplant at first relapse are both acceptable strategies in standard-risk patients
Non-Transplant Candidates
- VRd (6-9 months) followed by lenalidomide maintenance, OR
- DRd (daratumumab + lenalidomide + dexamethasone) until progression
Relapsed/Refractory Myeloma
Key drug classes and specific agents:
| Class | Agents |
|---|---|
| Proteasome inhibitors | Bortezomib, carfilzomib, ixazomib |
| Immunomodulatory drugs (IMiDs) | Thalidomide, lenalidomide, pomalidomide |
| Anti-CD38 monoclonal antibodies | Daratumumab, isatuximab |
| Anti-SLAMF7 antibody | Elotuzumab |
| CAR-T cell therapy | Ide-cel (idecabtagene vicleucel), cilta-cel |
| Bispecific antibodies | Teclistamab, elranatamab, talquetamab (target BCMA) |
| BCL-2 inhibitor | Venetoclax (for t(11;14) only) |
| Exportin-1 inhibitor | Selinexor |
Mechanism of proteasome inhibitors: Myeloma cells have high immunoglobulin synthesis with intrinsic protein misfolding. Blocking proteasomal degradation causes accumulation of misfolded proteins, triggering apoptosis selectively in these cells.
Complication Management
- Hypercalcemia: IV hydration + steroids + bisphosphonates (zoledronic acid or pamidronate)
- Renal failure (myeloma kidney): Aggressive hydration, furosemide (maintain urine output >100 mL/hr), plasmapheresis if serum FLC >150 mg/dL, bortezomib-based regimen
- Bone disease: Bisphosphonates for all patients with bone lesions; surgical fixation for pathologic fractures
- Infections: Prompt antibiotics; consider prophylactic IVIG in recurrent infections
- Radiation: Palliative 20-30 Gy for focal unresponsive pain or spinal cord compression
Prognosis
- 5-year survival ~60% overall
- Untreated multi-lesion disease: survival <6-12 months
- "Smoldering myeloma": may remain asymptomatic for years
- Outcomes have improved substantially over the past 2 decades with proteasome inhibitors, IMiDs, anti-CD38 antibodies, and CAR-T therapies
Spectrum of Plasma Cell Disorders
| Condition | Key Feature |
|---|---|
| MGUS | M protein <3 g/dL, <10% marrow plasma cells, no CRAB; no treatment needed |
| Smoldering MM | M protein ≥3 g/dL or ≥10% plasma cells but no CRAB criteria |
| Multiple Myeloma | CRAB criteria + clonal plasma cells |
| Solitary plasmacytoma | Single lesion; <10% marrow plasma cells elsewhere |
| Plasma cell leukemia | >20% plasma cells in peripheral blood |
Sources: Goldman-Cecil Medicine; Robbins & Kumar Pathologic Basis of Disease (10th ed.); Robbins, Cotran & Kumar Pathologic Basis of Disease; Campbell's Operative Orthopaedics 15e
acute chest syndrome in sickle cell anemia
Finding Sources
Searching PubMed
"acute chest syndrome"[MeSH Terms] AND "sickle cell"[MeSH Terms]
Finding Sources
Searching PubMed
"acute chest syndrome" AND "sickle cell anemia"
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
I now have comprehensive information from multiple authoritative textbooks. Here is the full answer:
Acute Chest Syndrome (ACS) in Sickle Cell Anemia
Definition
ACS is defined as a new pulmonary infiltrate on chest radiograph plus at least one of the following new signs or symptoms:
- Fever >38.5°C (101.3°F)
- Cough or wheezing
- Tachypnea
- Chest pain
- Hypoxia
It is the leading cause of death in patients with HbSS disease in the United States, with mortality up to 9%, and up to 13% of patients require mechanical ventilation. - Tintinalli's Emergency Medicine
Pathogenesis
ACS arises from a self-reinforcing vicious cycle. Multiple triggers converge to produce regional hypoxia, lung injury, and worsening sickling:
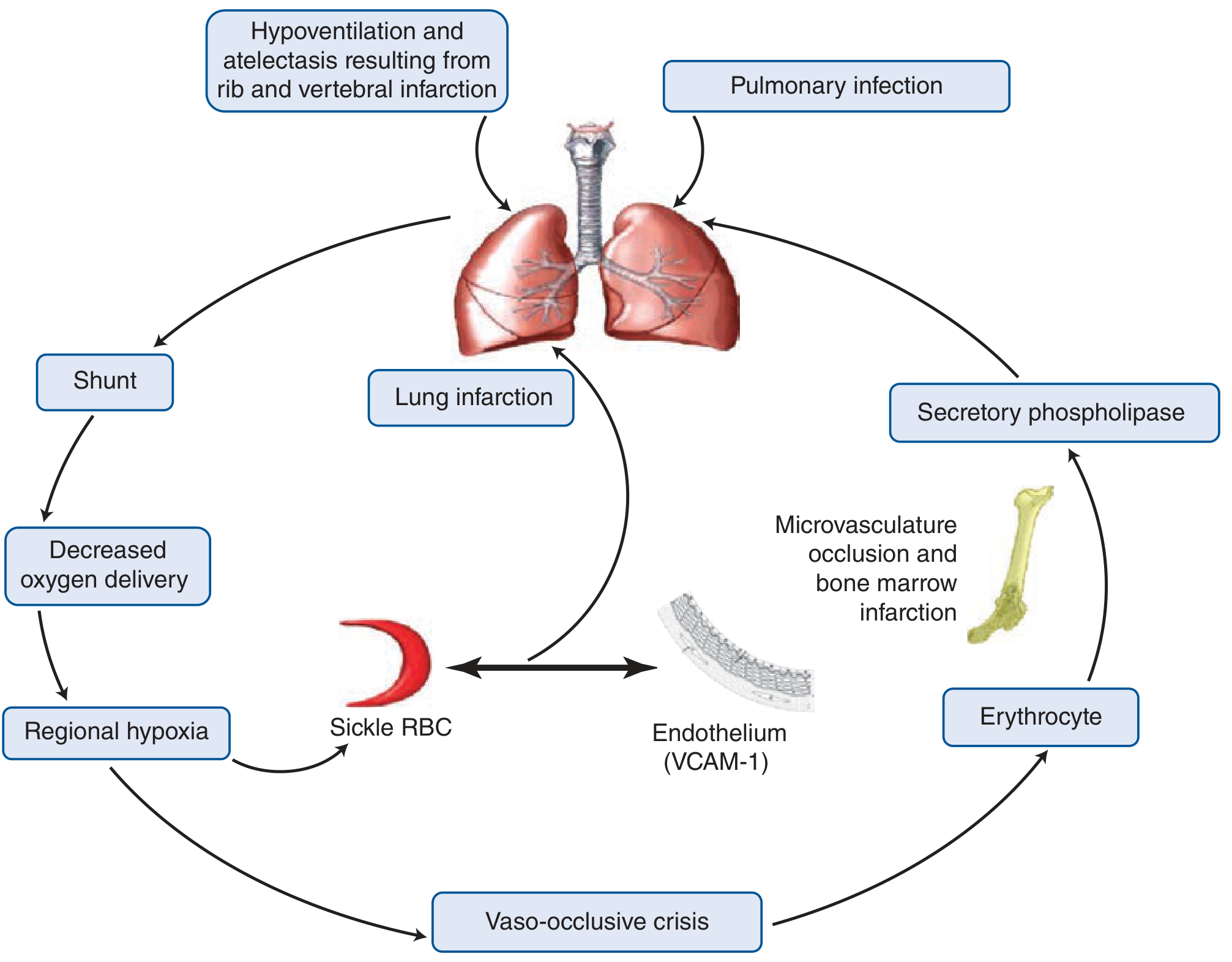
Key mechanisms:
1. In situ sickling within the pulmonary vasculature
- Sluggish blood flow in an inflamed or infected lung bed causes deoxygenation and local HbS polymerization
- This worsens pulmonary and systemic hypoxemia, causing further sickling - a vicious cycle
2. Fat embolism from infarcted bone marrow
- Bony slivers and marrow fat found in pulmonary vasculature at autopsy
- Fat droplets within endothelial cells identified on bronchoscopy in living patients
- Elevated serum free fatty acids and secretory phospholipase A2 (potent inflammatory mediator from bone marrow) are biomarkers
3. Pulmonary infection
- Up to 30% of ACS cases have an identifiable infectious pathogen
- Most common: Chlamydia pneumoniae (adults) and Mycoplasma pneumoniae (children)
- Others: Staphylococcus aureus, Haemophilus influenzae, Klebsiella pneumoniae, adenovirus, influenza, RSV, parvovirus B19
- Note: Streptococcus pneumoniae was historically common but is now rare due to pneumococcal immunization and penicillin prophylaxis
4. Hypoventilation-induced atelectasis
- Rib and vertebral infarction causes splinting and hypoventilation
- Opioid analgesia used for pain crises can worsen hypoventilation
5. Pulmonary thrombosis
- Pulmonary thrombosis has high prevalence in sickle cell anemia and is present in ~17% of ACS cases
Timing: In adults, ACS most commonly occurs 1 to 3 days after hospitalization for an acute pain crisis. - Tintinalli's Emergency Medicine
Clinical Features
| Feature | Frequency |
|---|---|
| Fever | ~80% |
| Cough | up to 74% |
| Chest pain (often pleuritic) | 44-57% |
| Rales on auscultation | 48-76% (most common exam finding) |
| Wheezing/bronchospasm | present in some; may persist |
Bronchial hyperactivity can persist after recovery, suggesting ACS causes lasting lung injury in some patients.
Investigations
- Chest X-ray: Required - new infiltrate (may be single or multilobar). In severe disease may progress rapidly
- ABG / pulse oximetry: Assess degree of hypoxemia; PaO2 <60 mmHg signals severe disease
- CBC: Anemia, leukocytosis
- Blood cultures + sputum/BAL cultures: Identify organisms
- Serum secretory phospholipase A2: Elevated; predicts impending ACS
- Bone scan / MRI: Distinguish rib infarction from infection if unclear
Treatment
Treatment is supportive combined with antibiotics and transfusion:
1. Oxygen
- Supplement for all hypoxemic patients
- Target SaO2 >95%
2. Analgesics
- Parenteral opioids for chest/bone pain
- Careful monitoring essential - opioid-induced hypoventilation can worsen ACS
- Incentive spirometry recommended (both treatment and prevention)
3. Fluids
- IV maintenance fluids (5% dextrose in half normal saline) until oral intake is adequate
- Avoid volume overload - may worsen pulmonary edema
4. Antibiotics
- Empirical broad-spectrum antibiotics recommended irrespective of culture results
- Standard regimen: parenteral cephalosporin + macrolide (covers Mycoplasma and Chlamydia)
- Inhaled beta-2 agonists if wheezing is present
5. Transfusion
- Considered lifesaving, though no firm RCT-based indications exist
| Transfusion Type | Indication |
|---|---|
| Simple transfusion | Mild-moderate ACS; Hgb <9 g/dL |
| Exchange transfusion | Severe ACS (PaO2 <60 mmHg), neurologic complications, multiorgan failure, or relatively high Hgb (>9 g/dL) |
- Exchange transfusion goal: reduce HbS to <30%
- Exchange transfusion is preferred when Hgb is already high (>9 g/dL) because simple transfusion increases viscosity and risks complications
- Exchange transfusion decreases HbS concentration without iron gain and maintains stable whole-blood viscosity
6. Corticosteroids
- Controversial; may prevent deterioration and reduce transfusion need but are associated with higher readmission rates
7. Respiratory support
- ICU admission for severe cases
- Noninvasive (CPAP/BiPAP) or invasive ventilation as needed
Disposition and Prognosis
- All patients with ACS must be admitted to a monitored setting
- Predictors of poor outcome:
- Older age
- Platelets <199,000/mm³
- Comorbid cardiac disease
- Extensive/multilobar lung involvement
- Long-term complications: Pulmonary hypertension, pulmonary fibrosis, cor pulmonale
Although most patients have single episodes, recurrent ACS leads to chronic sickle lung disease. ACS and stroke are the two leading causes of ischemia-related death in sickle cell disease. - Robbins & Kumar Basic Pathology
Prevention
Hydroxyurea is the cornerstone of long-term prevention:
- Reduces the rate of pain crises and ACS episodes by approximately 50% - Symptom to Diagnosis, 4th Ed.
- Mechanism: Increases HbF (which does not polymerize), reduces leukocyte count (anti-inflammatory), increases RBC volume (reduces intracellular HbS concentration)
- Indications for hydroxyurea: All infants, children, and adolescents with sickle cell anemia (regardless of severity); adults with recurrent pain, severe or recurrent ACS, or symptomatic anemia - Goldman-Cecil Medicine
Additional preventive measures:
- Pneumococcal, meningococcal, and Haemophilus influenzae vaccinations
- Prophylactic penicillin (especially in children <5 years)
- Chronic transfusion therapy in selected high-risk patients
- Newer agents: Voxelotor (prevents HbS polymerization), Crizanlizumab (anti-P-selectin, reduces vaso-occlusion), which can be combined with hydroxyurea
Sources: Tintinalli's Emergency Medicine; Goldman-Cecil Medicine; Robbins & Kumar Basic Pathology; Quick Compendium of Clinical Pathology 5e
This is a shared conversation. Sign in to Orris to start your own chat.