Explain Inflammation in depth, acute and chronic in east language w.r.t. Robbins and Cotran Pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive, easy-language explanation. Let me compile it.
Inflammation — A Complete Guide
Based on Robbins, Cotran & Kumar: Pathologic Basis of Disease
🔷 What Is Inflammation?
Inflammation is the response of vascularized tissues that delivers leukocytes and defense molecules from the blood to sites of infection or cell damage. Despite the negative connotation the word carries in everyday life, inflammation is fundamentally protective — without it, infections would go unchecked, wounds would never heal, and injured tissue would become a "permanent festering sore."
Think of it like the body's 911 system — it detects a problem, calls in reinforcements, eliminates the threat, and then tries to repair the damage.
The suffix -itis appended to an organ name = inflammation of that organ (e.g., appendicitis, meningitis, hepatitis).
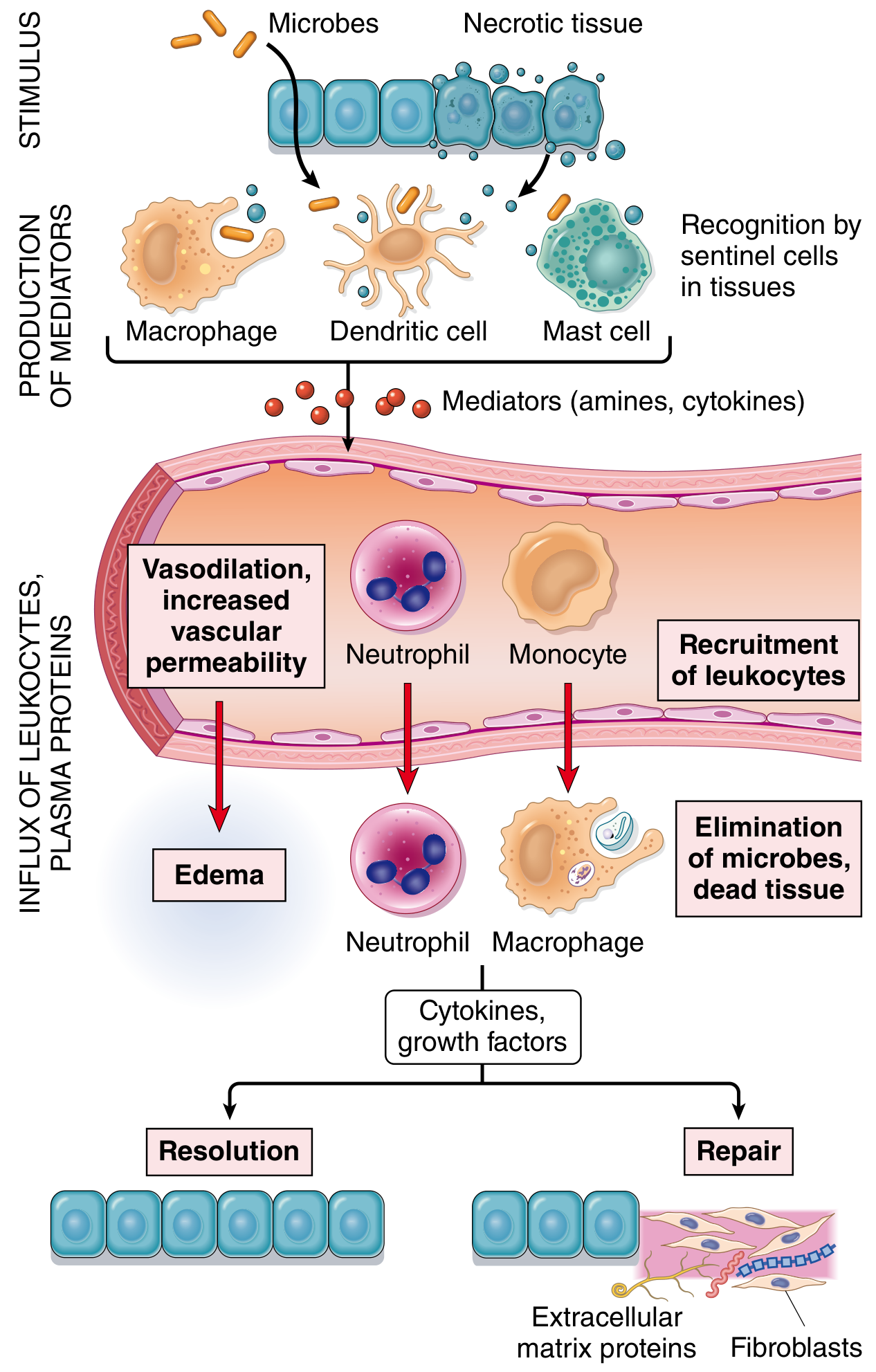
🔷 Causes of Inflammation
| Category | Examples |
|---|---|
| Infections | Bacteria, viruses, fungi, parasites |
| Tissue necrosis | Ischemia/infarction, trauma, chemical injury |
| Foreign bodies | Splinters, sutures, silica dust |
| Immune reactions | Autoimmune diseases, hypersensitivity reactions |
Cells detect injury/infection through pattern-recognition receptors (PRRs), especially Toll-like receptors (TLRs) on macrophages and dendritic cells. These recognize:
- PAMPs — Pathogen-Associated Molecular Patterns (bacterial lipopolysaccharide, viral dsRNA)
- DAMPs — Damage-Associated Molecular Patterns (ATP, uric acid, HMGB1 released from dying cells)
PART 1 — ACUTE INFLAMMATION
🔸 Definition
Acute inflammation is a rapid, short-lived response (minutes to days) characterized by:
- Vascular dilation → increased blood flow
- Increased vascular permeability → plasma proteins exit into tissues
- Leukocyte (mainly neutrophil) emigration → accumulate and destroy the offending agent
🔸 The Five Cardinal Signs (Celsus + Virchow)
| Latin | English | Mechanism |
|---|---|---|
| Rubor | Redness | Vasodilation → more blood |
| Calor | Heat | Increased blood flow (+ systemic fever) |
| Tumor | Swelling | Fluid exudate leaks into tissue |
| Dolor | Pain | Prostaglandins, bradykinin, substance P stimulate pain fibers |
| Functio laesa | Loss of function | Pain + tissue damage |
🔸 Step 1 — Vascular Reactions
A. Changes in Flow & Caliber
After injury, blood vessels go through a rapid sequence:
- Transient vasoconstriction — lasts only seconds
- Vasodilation (mainly postcapillary venules) — driven by histamine → redness + heat
- Increased permeability — plasma proteins pour into tissues → exudate forms → swelling
- Stasis — blood slows as fluid is lost; red cells concentrate → vessel engorgement
- Leukocytes line up (margination) along activated endothelium
B. Exudate vs. Transudate
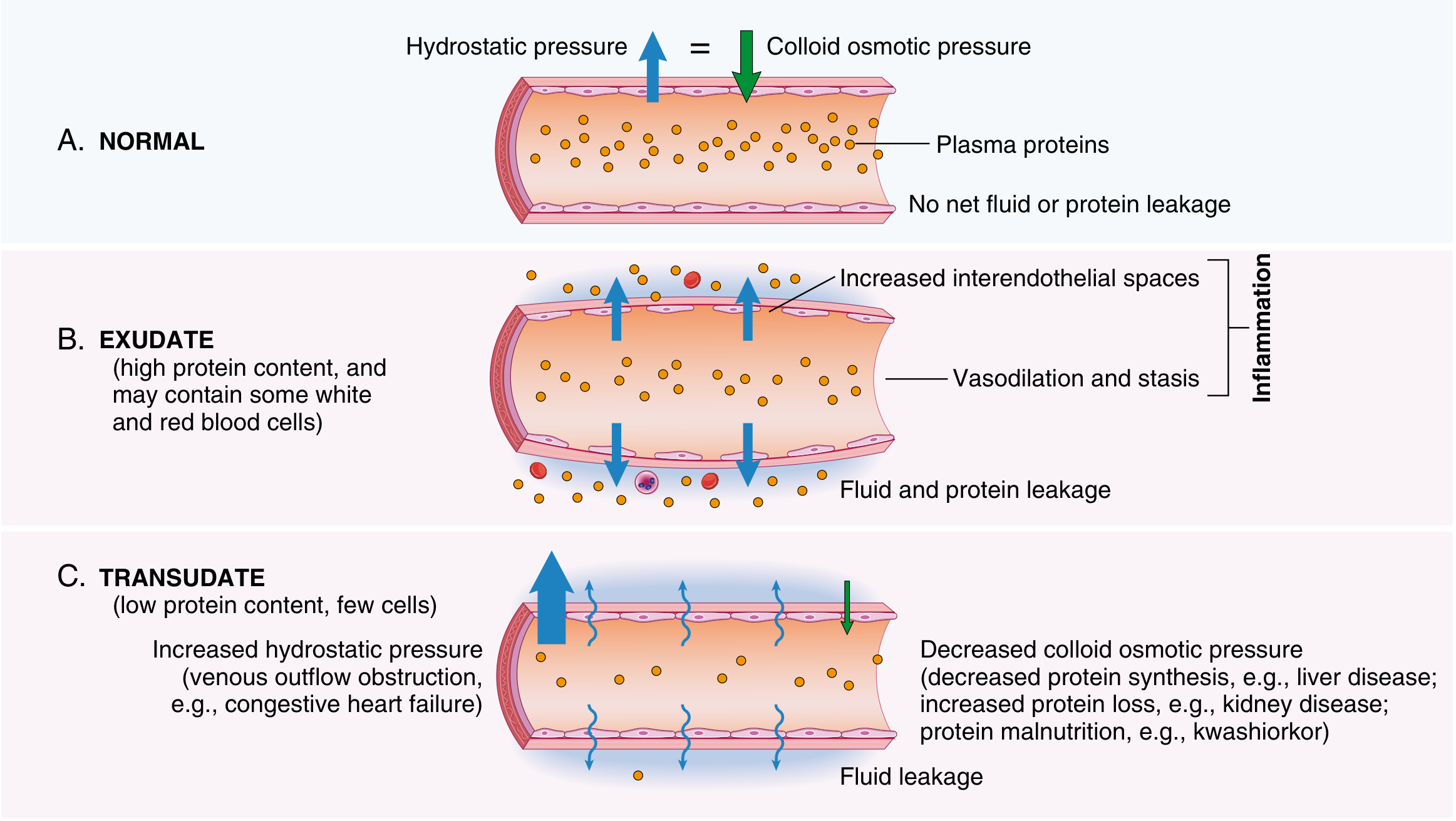
| Exudate | Transudate | |
|---|---|---|
| Protein | High | Low (mostly albumin) |
| Cells | Many leukocytes | Few/none |
| Cause | Inflammation (increased permeability) | Heart failure, nephrotic syndrome |
| Pus | Purulent exudate (neutrophils + debris + microbes) | — |
🔸 Step 2 — Leukocyte Recruitment
This is the cellular reaction of acute inflammation — getting neutrophils from blood to the injury site. It proceeds in sequential steps:
🅐 Margination & Rolling
- With stasis, neutrophils drift to the vessel periphery (margination)
- They loosely roll along activated endothelium via selectins (P-selectin, E-selectin on endothelium binding sialyl-Lewis X on leukocytes)
🅑 Adhesion (Firm Arrest)
- Chemokines activate leukocytes → upregulate integrins (LFA-1, MAC-1)
- Integrins bind ICAM-1 on endothelium → firm adhesion
🅒 Transmigration (Diapedesis)
- Leukocytes squeeze between endothelial cells (through tight junctions) by binding PECAM-1 (CD31)
- They then cross the basement membrane using collagenases
🅓 Chemotaxis
- Leukocytes migrate along a chemical gradient toward the injury site
- Key chemoattractants: C5a (complement), LTB₄ (leukotriene B4), IL-8/CXCL8 (chemokine), bacterial products (fMLP)
Simple memory trick: Roll → Stick → Squeeze through → Crawl toward the problem (RSSC)
🔸 Step 3 — Phagocytosis
Once neutrophils (and later macrophages) arrive, they eliminate the offending agent in 3 steps:
1. Recognition & Attachment
- Phagocytes use opsonins to boost binding — IgG (via Fc receptor), C3b (via complement receptor), mannose-binding lectin
- Opsonization = coating a target to make it easier to eat
2. Engulfment
- Cytoplasmic extensions (pseudopods) surround the particle → form a phagosome → fuses with lysosome → phagolysosome
3. Killing
- Reactive Oxygen Species (ROS): NADPH oxidase generates superoxide (O₂⁻) → H₂O₂ → hypochlorous acid (HOCl, the most potent) in the "respiratory burst"
- Reactive Nitrogen Species (RNS): iNOS generates nitric oxide (NO) → reacts with O₂⁻ → peroxynitrite (ONOO⁻)
- Lysosomal enzymes: Proteases, elastase, myeloperoxidase, defensins
Chediak-Higashi syndrome = defective lysosome-phagosome fusion → recurrent infections Chronic Granulomatous Disease (CGD) = NADPH oxidase defect → no respiratory burst → catalase-positive organisms survive
Neutrophil Extracellular Traps (NETs)
Neutrophils can also expel their own DNA + histones + antimicrobial proteins as a sticky web to trap and kill extracellular microbes. This is a specialized killing mechanism that sacrifices the neutrophil.
🔸 Mediators of Acute Inflammation
These are the "chemical messengers" that orchestrate the whole response.
Cell-Derived Mediators
| Mediator | Source | Actions |
|---|---|---|
| Histamine | Mast cells, basophils, platelets | Vasodilation, ↑ vascular permeability, endothelial activation |
| Prostaglandins (PGs) | Mast cells, leukocytes (via COX pathway) | Vasodilation, fever, pain |
| Leukotrienes (LTs) | Mast cells, leukocytes (via 5-LOX pathway) | ↑ permeability (LTC₄, D₄, E₄), chemotaxis (LTB₄) |
| TNF, IL-1, IL-6 | Macrophages, endothelium, mast cells | Endothelial activation, fever, acute phase response |
| Chemokines (IL-8) | Leukocytes, macrophages | Chemotaxis, leukocyte activation |
| PAF (Platelet-Activating Factor) | Leukocytes, mast cells | Vasodilation, ↑ permeability, platelet aggregation |
Plasma-Derived Mediators
| Mediator | System | Key Actions |
|---|---|---|
| C3a, C5a | Complement | Mast cell degranulation, chemotaxis |
| C5b–9 (MAC) | Complement | Direct cell lysis |
| Bradykinin | Kinin system | ↑ permeability, pain, vasodilation |
| Fibrin/thrombin | Coagulation | Clot formation, inflammation amplification |
NSAIDs (aspirin, ibuprofen) inhibit COX enzymes → block prostaglandin synthesis → reduce fever, pain, inflammation Corticosteroids inhibit phospholipase A₂ → block both prostaglandin AND leukotriene synthesis
🔸 Morphologic Patterns of Acute Inflammation
| Pattern | Characteristics | Example |
|---|---|---|
| Serous | Watery, protein-poor fluid; no/few cells | Skin blister (burn/herpes), pleural effusion |
| Fibrinous | Large fibrin deposits; large molecule leak | Pericarditis ("bread and butter" pattern), lobar pneumonia |
| Purulent (Suppurative) | Pus = neutrophils + debris + microbes | Abscess, furuncle, bacterial meningitis |
| Ulcer | Epithelial defect from acute/chronic inflammation | Peptic ulcer, aphthous ulcer |
🔸 Outcomes of Acute Inflammation
Three possible fates (Robbins Fig. 3.16):
Acute Inflammation
│
┌────┴──────────────────┐
▼ ▼
Complete Resolution Cannot Resolve
(short injury, minimal ────────────────────────
tissue destruction) │ │
Macrophages clear debris ▼ ▼
Lymphatics resorb fluid Scarring Chronic
Tissue regenerates (fibrosis) Inflammation
- Complete resolution — The ideal outcome. Debris cleared, edema resorbed, tissue regenerated back to normal.
- Healing by connective tissue (scarring/fibrosis) — When tissue can't regenerate (e.g., heart muscle, neurons) or when fibrin can't be cleared.
- Progression to chronic inflammation — When the injurious agent persists or healing is impaired.
PART 2 — CHRONIC INFLAMMATION
🔸 Definition
Chronic inflammation is a prolonged response (weeks to months) in which inflammation, tissue injury, and repair all occur simultaneously. It may follow acute inflammation or begin insidiously without an obvious acute phase.
🔸 Causes of Chronic Inflammation
| Setting | Mechanism | Example |
|---|---|---|
| Persistent infections | Organisms resist eradication → T-cell hypersensitivity | Tuberculosis (MTB), Helicobacter pylori, hepatitis C, schistosomiasis |
| Immune/hypersensitivity diseases | Auto-antigens or harmless antigens trigger self-perpetuating reaction | Rheumatoid arthritis, MS, IBD (Crohn's), asthma |
| Toxic agents | Nondegradable material → persistent macrophage activation | Silicosis (silica dust), atherosclerosis (oxidized LDL) |
🔸 Morphologic Features of Chronic Inflammation
Unlike acute inflammation (neutrophils, edema), chronic inflammation is defined by three simultaneous processes:
- Mononuclear cell infiltrate — macrophages, lymphocytes, and plasma cells (NOT neutrophils)
- Tissue destruction — caused by the persistent offending agent AND by the inflammatory cells themselves
- Repair — angiogenesis + fibrosis trying to fill the damage
The battlefield never clears — fighting and rebuilding happen at the same time.
🔸 Key Cells in Chronic Inflammation
1. Macrophages — The Central Generals
Macrophages are the dominant cells of chronic inflammation. They originate from:
- Circulating monocytes (from bone marrow)
- Tissue-resident macrophages seeded from yolk sac/fetal liver before birth (Kupffer cells in liver, microglia in brain, alveolar macrophages in lung)
Activation states:
- M1 (Classical activation) — by IFN-γ and microbial products → produce ROS, NO, TNF, IL-1, IL-12 → kill microbes → pro-inflammatory
- M2 (Alternative activation) — by IL-4, IL-13 (from Th2 cells) → secrete IL-10, TGF-β → tissue repair, fibrosis, anti-inflammatory
Macrophages drive chronic inflammation by:
- Secreting cytokines (TNF, IL-1, IL-12) that perpetuate the response
- Presenting antigens to T lymphocytes
- Producing growth factors that stimulate fibrosis (TGF-β, PDGF)
2. Lymphocytes — The Memory Soldiers
- CD4+ T helper cells are the main lymphocytes in chronic inflammation
- Th1 cells produce IFN-γ → activate macrophages (classical M1 activation)
- Th2 cells produce IL-4, IL-5, IL-13 → activate eosinophils + alternative macrophage activation
- Th17 cells produce IL-17 → recruit neutrophils
- CD8+ cytotoxic T cells kill infected cells directly
- Macrophage–T cell crosstalk forms a self-amplifying loop in diseases like TB and sarcoidosis
3. Other Cells
| Cell | When prominent |
|---|---|
| Plasma cells | Antibody production in persistent antigenic stimulation |
| Eosinophils | Parasitic infections, allergic reactions (IgE-mediated) |
| Mast cells | Allergic inflammation; found in connective tissues throughout body |
🔸 Granulomatous Inflammation — A Special Pattern
This is the most important pattern of chronic inflammation to know.
Definition: A granuloma is a focal area of granulomatous inflammation consisting of clusters of activated macrophages (called epithelioid cells because they look like epithelial cells) often surrounded by a collar of lymphocytes, sometimes with multinucleated giant cells.
Two types of granulomas:
| Feature | Immune Granuloma | Foreign Body Granuloma |
|---|---|---|
| Cause | Insoluble antigen triggering T-cell response | Inert material (suture, silica, talc) |
| Giant cells | Langhans-type (nuclei at periphery) | Foreign body-type (nuclei scattered) |
| T cells | Present (CD4+, IFN-γ-producing) | Absent |
| Caseation | Possible (in TB) | Absent |
Classic causes to remember:
- TB — caseating granuloma (central cheesy necrosis) + Langhans giant cells
- Sarcoidosis — non-caseating granuloma (no necrosis)
- Crohn's disease — non-caseating granuloma
- Leprosy, Cat-scratch disease, Schistosomiasis, Fungal infections
Granuloma = body's way of "walling off" what it cannot destroy
🔸 Systemic Effects of Inflammation ("Acute Phase Response")
When inflammation is severe, local mediators (TNF, IL-1, IL-6) enter the blood and cause systemic effects:
| Systemic Effect | Mediator | Mechanism |
|---|---|---|
| Fever | IL-1, TNF, IL-6 → prostaglandins (PGE₂) | Act on hypothalamus → reset thermostat |
| Leukocytosis | IL-1, TNF, CSFs | Stimulate bone marrow → release more WBCs |
| Elevated ESR & CRP | IL-6 → liver | Acute phase proteins (CRP, fibrinogen, haptoglobin) rise |
| Hypotension/shock | TNF | Widespread vasodilation |
| Anemia of chronic disease | Hepcidin (via IL-6) | Iron sequestration |
| Fatigue, anorexia | IL-1, TNF, IL-6 | Act on hypothalamus/brain |
CRP (C-reactive protein) is produced by the liver in response to IL-6 and is a widely used clinical marker of inflammation. It binds phosphocholine on microbes and dead cells, acting as an opsonin.
🔷 Acute vs. Chronic Inflammation — Quick Comparison
| Feature | Acute | Chronic |
|---|---|---|
| Onset | Minutes to hours | Weeks to months |
| Duration | Short | Prolonged |
| Main cells | Neutrophils | Macrophages, lymphocytes, plasma cells |
| Edema | Prominent | Less prominent |
| Tissue injury | Usually mild | Usually significant + ongoing |
| Fibrosis | No | Often present |
| Vascular changes | Prominent (vasodilation, ↑ permeability) | Less prominent |
| Outcome | Resolution, scar, or → chronic | Fibrosis, tissue destruction, sometimes resolution |
| Examples | Appendicitis (early), bacterial pneumonia, blister | TB, RA, Crohn's, silicosis |
🔷 Key Takeaway (Robbins Summary)
Inflammation is a double-edged sword — it is essential for host defense and healing, but when it goes unchecked, it becomes the engine of widespread tissue destruction. Virtually all chronic diseases — atherosclerosis, type 2 diabetes, cancer, Alzheimer's, autoimmune disorders — have inflammation as a central driver.
The art of modern medicine is learning how to harness inflammation when needed (vaccines, wound healing) and suppress it when it becomes the disease (steroids, biologics targeting TNF/IL-1/IL-6).
— Robbins, Cotran & Kumar: Pathologic Basis of Disease, Chapter 3: Inflammation and Repair
Also explain healing and scarring mechanism in easy language as per Robbins and Cotran pathology
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.