Ewing sarcoma
Reading File
Finding Sources
Searching PubMed
"Ewing sarcoma"[MeSH Terms] AND treatment
Searching the Web
Ewing sarcoma 2025 clinical overview diagnosis treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Searching PubMed
"Ewing sarcoma"[MeSH Terms] AND chemotherapy
Reading File
Loading Image
Reading File
I now have comprehensive information to write a thorough response. Let me compile everything.
Ewing Sarcoma
Epidemiology
Ewing sarcoma is the third most common primary malignant bone tumor overall, but ranks second after osteosarcoma in patients under 30, and is the most common primary bone malignancy in children under 10 years. Incidence is less than 1 per million per year; roughly 200 cases are diagnosed in the United States annually.
-
Age: typically 5-25 years (peak: second decade); ~20% occur in older patients
-
Sex: slight male predominance
-
Race: exceedingly rare in individuals of African descent - predominantly affects people of European origin
-
No known predisposing factors
-
Campbell's Operative Orthopaedics, 15th Ed. 2026, p. 1221
-
Robbins & Kumar Basic Pathology, p. 786
Pathogenesis & Molecular Biology
Ewing sarcoma belongs to the small round blue cell tumor family. Its molecular hallmark is a chromosomal translocation involving the EWSR1 gene on chromosome 22:
| Translocation | Fusion Gene | Frequency |
|---|---|---|
| t(11;22)(q24;q12) | EWSR1-FLI1 | ~85% |
| t(21;22)(q22;q12) | EWSR1-ERG | ~10% |
| t(7;22)(p22;q12) | EWSR1-ETV1 | rare |
The chimeric EWS/FLI1 protein binds chromatin and dysregulates transcription, driving uncontrolled growth and abnormal differentiation. The cell of origin remains uncertain - mesenchymal stem cells and primitive neuroectodermal cells are the leading candidates.
- Robbins & Kumar Basic Pathology, p. 786
- Goldman-Cecil Medicine, p. 2114
Sites of Origin
Common sites:
- Diaphysis (classically) and metaphysis (more frequent in practice) of long bones, often with extensive diaphyseal extension
- Flat bones: pelvis and ribs (large proportion of cases)
- Femur, tibia, fibula, humerus
Less common:
- Vertebral column (3.3-15% of cases; sacrum involved in up to 50% of spinal cases)
- Small bones of hands and feet (rare)
- Extraskeletal (extraosseous) Ewing sarcoma: ~20% of cases arise entirely in soft tissue
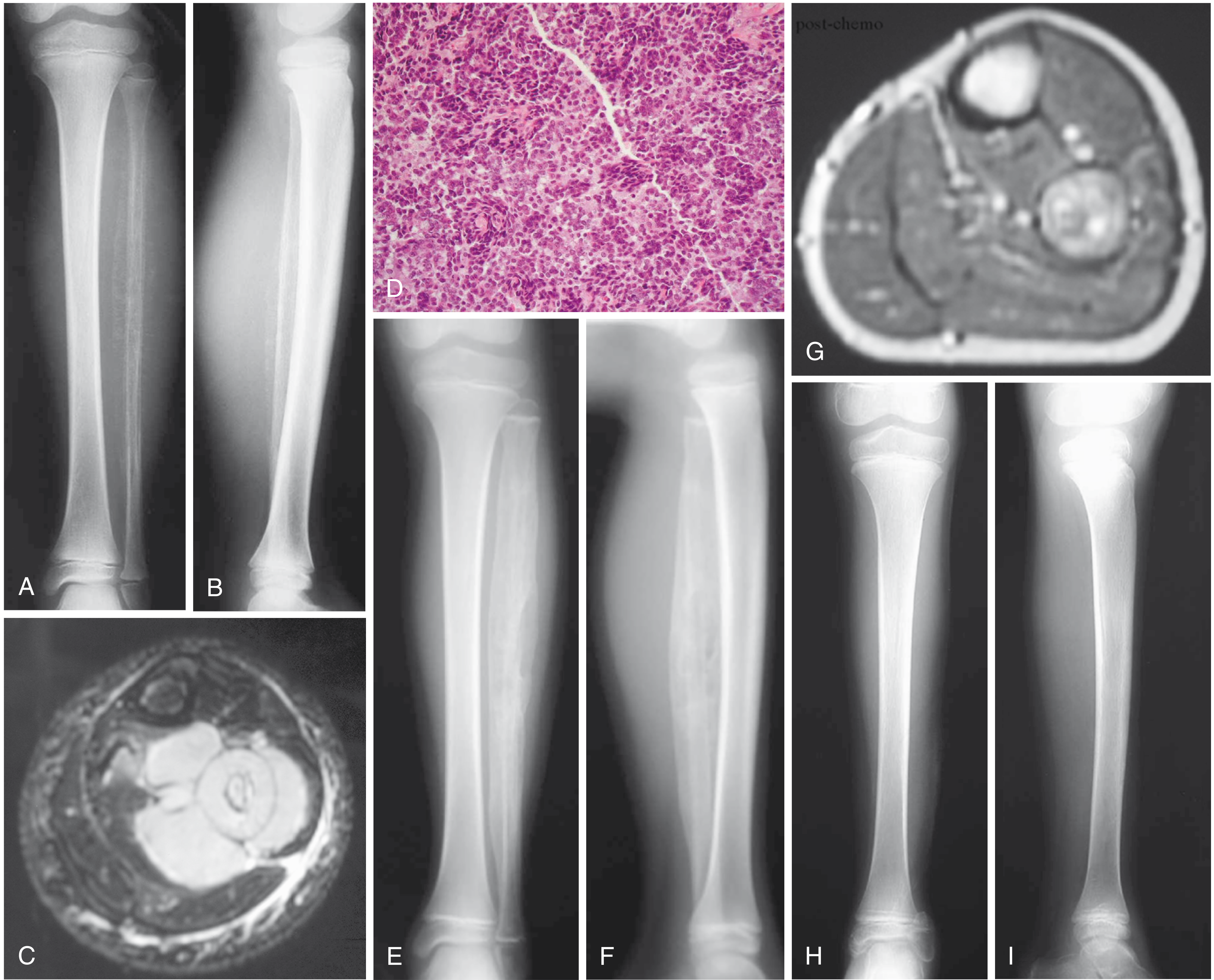
Clinical Presentation
- Pain - nearly universal, often insidious onset with long delay before diagnosis
- Average delay: 34 weeks from symptom onset to diagnosis
- Patient delay: ~15 weeks; physician delay: ~19 weeks
- Soft-tissue mass - often large at presentation
- Fever, erythema, swelling - can mimic osteomyelitis (a key diagnostic pitfall)
- Systemic signs: intermittent low-grade fever, leukocytosis, anemia, elevated ESR and CRP
- Important: a needle aspirate may grossly resemble pus - always send specimens to both microbiology AND pathology
Imaging
Plain radiography:
- Destructive "permeative" or "moth-eaten" lesion
- Classic "onion-skin" periosteal reaction (multilayered)
- Typically diaphyseal, though metaphyseal origin is common
- Often involves a large portion or the entire bone
- Flat bones: nonspecific destructive lesion
MRI (study of choice for local staging):
- Must image the entire bone to assess full extent (often extends beyond plain film abnormality)
- Isointense on T1, isointense-to-hyperintense on T2
- Intense gadolinium enhancement (hypercellular tumor)
- Frequently shows large paraspinal/soft-tissue component
- Assesses neurovascular involvement
Staging workup:
- CXR and chest CT (lung = most common metastatic site)
- Bone scan (bone = second most common metastatic site)
- FDG-PET/CT: now standard for initial staging, detection of recurrence; initial SUV correlates with tumor aggressiveness
- Bone marrow aspirate (at some institutions) or whole-body MRI to exclude diffuse systemic disease
Histology & Pathology
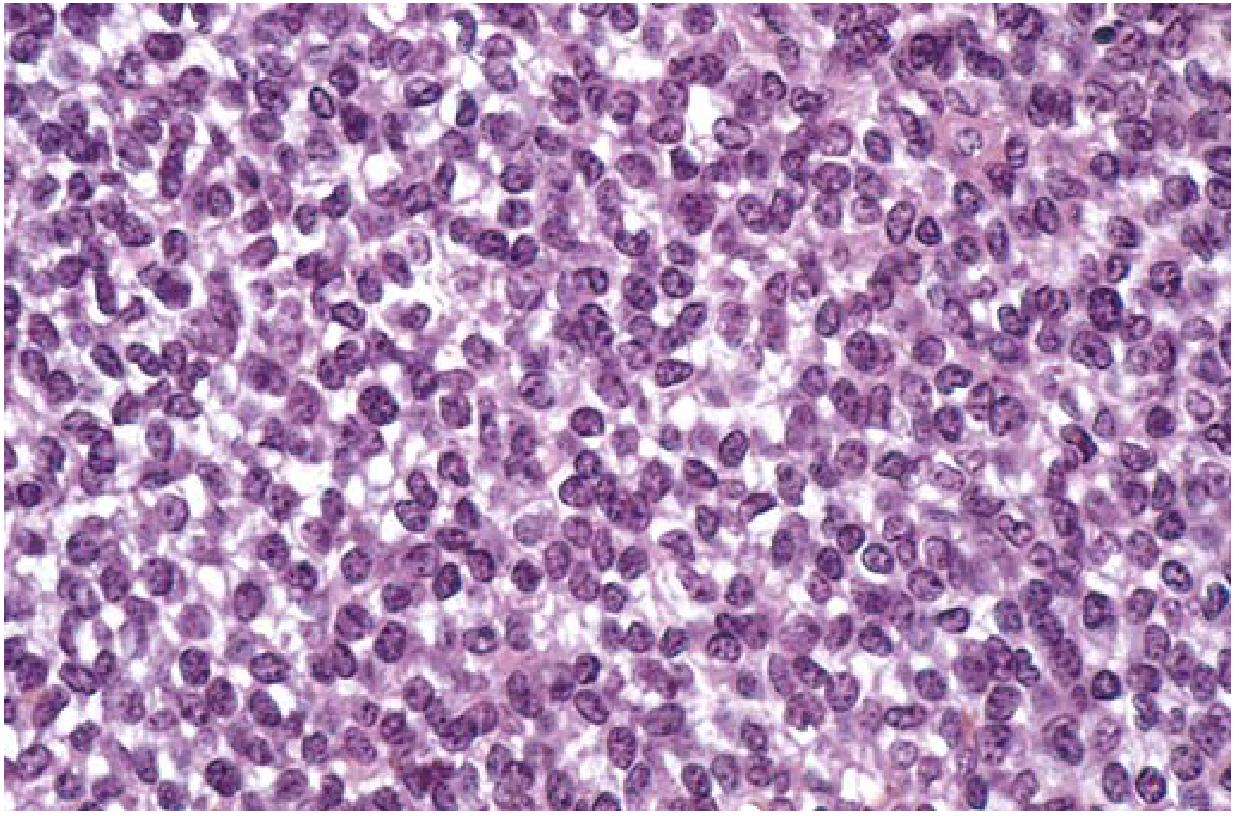
Microscopically, Ewing sarcoma shows:
- Sheets of uniform small round cells with scant cytoplasm (slightly larger than lymphocytes)
- Cytoplasm may appear clear due to intracellular glycogen (PAS-positive, reticulin-negative)
- Homer-Wright rosettes may be present (circular cell groupings with central fibrillary core)
- Tumor cells do NOT produce bone or cartilage
Immunohistochemistry for differential diagnosis:
| Marker | Ewing | Lymphoma | Rhabdomyosarcoma |
|---|---|---|---|
| PAS | + | - | - |
| Reticulin | - | + | variable |
| LCA / T-B cell markers | - | + | - |
| MIC-2 (CD99) | + | - | - |
| Desmin/myoglobin | - | - | + |
MIC-2 (CD99) immunostaining is considered specific for Ewing sarcoma and is the key immunohistochemical marker.
Treatment
Treatment requires a multimodal approach - chemotherapy is mandatory because Ewing sarcoma is considered a systemic disease from the outset.
1. Neoadjuvant Chemotherapy
Standard regimen: VDC/IE - dose-intensive Vincristine, Doxorubicin, Cyclophosphamide alternating with Ifosfamide and Etoposide. This is administered before local treatment to:
- Reduce tumor size/soft-tissue component
- Treat micrometastatic disease
- Allow reassessment of surgical options
2. Local Treatment (Surgery vs. Radiation)
This is the most controversial aspect of management - decided case-by-case:
Surgery (preferred when feasible):
- Wide resection with negative margins
- Reported local recurrence rate <10% with wide resection
- Limb salvage surgery preferred; amputation if necessary
- After neoadjuvant chemotherapy, repeat staging (X-ray often shows increased ossification; MRI shows decreased soft-tissue mass)
Radiation:
- Ewing sarcoma is radiosensitive
- Used when wide margins are unachievable or functional deficit from surgery would be unacceptable
- Also used as adjuvant after marginal or contaminated resection
- Tumors in pelvis or spine often treated with chemoradiation due to surgical morbidity
3. Adjuvant Chemotherapy
Continued after local treatment to address systemic disease.
Salvage Regimens (relapsed/refractory):
- Cyclophosphamide + topotecan
- Irinotecan + temozolomide
- Gemcitabine + docetaxel
A 2025 phase Ib trial (Rego-Inter-Ewing-1) presented at ASCO 2025 tested regorafenib in combination with standard chemotherapy as first-line therapy for multi-metastatic Ewing sarcoma, showing promising results.
Prognosis
| Scenario | 5-Year Survival |
|---|---|
| Overall (localized disease) | 60-75% |
| With multiagent chemo (event-free) | ~70% |
| Metastatic disease at presentation | 20-30% |
| Local recurrence | ~20% |
| Distant metastatic relapse | ~10% |
Poor prognostic factors:
- Distant metastases (strongest negative factor)
- Large primary tumor size (>8 cm often cited)
- Pelvic/axial location
- Older age at diagnosis (~>12-15 years)
- Male sex
- Systemic symptoms (fever, anemia, elevated LDH/ESR/WBC)
- Poor histologic response to neoadjuvant chemotherapy (<90% necrosis is poor response; 100% necrosis = good response)
- Aberrant TP53 expression (secondary genetic alteration)
Note: All Ewing sarcomas are considered high grade by definition - histologic grade does not affect prognosis. The specific translocation subtype (t11;22 vs t21;22) also does not appear to affect clinical course.
Key Differentials (Small Round Blue Cell Tumors in Children)
- Ewing sarcoma - CD99+, PAS+, EWSR1 translocation
- Lymphoma - LCA+, PAS-, reticulin+
- Embryonal rhabdomyosarcoma - desmin+, myoglobin+, muscle-specific actin+
- Neuroblastoma - NSE+, catecholamines elevated
- Small cell osteosarcoma - produces osteoid
- Osteomyelitis - must always be excluded clinically
Sources:
- Campbell's Operative Orthopaedics, 15th Ed. 2026, pp. 1221-1224
- Robbins & Kumar Basic Pathology, pp. 786-787
- Goldman-Cecil Medicine, p. 2114
- Grainger & Allison's Diagnostic Radiology, p. 1285
- GEIS Clinical Practice Guidelines 2024
- ASCO 2025: Rego-Inter-Ewing-1 trial results
This is a shared conversation. Sign in to Orris to start your own chat.