Hey, so basically, a thrombotic stroke which occurs from the atherosclerotic block due to the block rupture and causing a thrombus where the subendothelial collagen is exposed and leads to the formation of thrombi. And the new this is like, it's not actually dependent, like it is made up of fibrin. So it's not easily liberated, it is degradable, but it takes some time. In contrast, the embolic stroke where it is the commonest of the embolic stroke is the heart where it is usually coming from the heart, its juice and involving some hemorrhagic infarct, so this can thrombolyze and the reentry of blood leads to hemorrhagic infarct. But I want to discuss more why thrombotic stroke takes time to lyse and why embolic stroke can lyse fast. Just an ultra high yield, very short 2 to 3 line important flow chart kind of thing, like an arrow language, not a sentence arrow language. And you can explain it in detail, but in the end, summary should be like that so that I can copy it on my notes.
Why Thrombotic Stroke Takes Long to Lyse vs. Embolic Stroke Lyses Fast
The Core Difference: Clot Composition
- Plaque rupture → subendothelial collagen exposed → platelet activation + aggregation (primary plug)
- High shear arterial environment → platelet-rich "white thrombus" forms
- Coagulation cascade activated → dense fibrin mesh cross-links the platelets
- The fibrin is heavily cross-linked (Factor XIIIa-stabilized) → compact, organized, resistant structure
- It forms in situ, grows over time, and becomes tightly adherent to the vessel wall
- Plasmin (the fibrinolytic enzyme) must penetrate a thick, organized, cross-linked fibrin network → slow lysis
- Thrombus forms in the heart under low shear / stasis conditions (e.g., AF, dilated LV, valvular disease)
- Low shear → fibrin-rich, platelet-poor "red thrombus" (lots of fibrin + trapped RBCs, fewer platelets)
- Less cross-linking, less compact, more accessible to plasmin
- Embolus travels distally → lodges in a cerebral artery
- Natural fibrinolysis (endogenous tPA from vessel wall) or exogenous thrombolytics can rapidly penetrate the loosely organized fibrin → fast lysis
- Rapid lysis → sudden reperfusion of ischemic, already-damaged capillaries → hemorrhagic transformation (the blood-brain barrier is broken → blood extravasates → hemorrhagic infarct)
Why Hemorrhagic Transformation in Embolic, Not Thrombotic?
| Factor | Thrombotic | Embolic |
|---|---|---|
| Lysis speed | Slow | Fast |
| Reperfusion | Gradual / incomplete | Sudden, brisk |
| BBB status at reperfusion | Partially recovers (slow ischemia) | Already severely damaged (dense ischemia in distal territory) |
| Hemorrhagic infarct | Rare | Common |
✦ Ultra High-Yield Flowchart (Arrow Language — Copy to Notes)
THROMBOTIC STROKE
Plaque rupture → subendothelial collagen exposed
→ Platelet-rich WHITE thrombus (high shear)
→ Dense, cross-linked fibrin mesh (Factor XIIIa-stabilized)
→ Adherent to vessel wall + organized structure
→ Plasmin penetration SLOW → lysis TAKES TIME
→ No sudden reperfusion → NO hemorrhagic infarct
──────────────────────────────────────────────────
EMBOLIC STROKE (Cardiac — commonest)
Cardiac stasis (AF/LV dysfunction)
→ Fibrin-rich RED thrombus (low shear, platelet-poor)
→ Loosely cross-linked → plasmin penetrates EASILY
→ Embolus lodges distally (end-artery, no collaterals)
→ Dense ischemia → BBB damaged
→ Clot lyses FAST → sudden reperfusion
→ Blood floods damaged capillaries → HEMORRHAGIC INFARCT
Sources: Braunwald's Heart Disease 11e (p. 2835); Bradley & Daroff's Neurology in Clinical Practice (block 8, line 594)
usmle topper level notes ultra super Hy usmle point of view
⚡ THROMBOTIC vs EMBOLIC STROKE — USMLE MASTER NOTES
🔴 THROMBOTIC STROKE
Atherosclerotic plaque (large vessel)
→ Plaque rupture → subendothelial COLLAGEN exposed
→ Platelet adhesion (GPIb–vWF) → activation → aggregation (GPIIb/IIIa–fibrinogen)
→ HIGH SHEAR environment → PLATELET-RICH "WHITE THROMBUS"
→ Coagulation cascade → DENSE, CROSS-LINKED fibrin (Factor XIIIa stabilized)
→ Thrombus IN SITU → adherent to vessel wall
→ Plasmin access BLOCKED (dense fibrin) → SLOW LYSIS
→ Gradual onset, stepwise worsening ("stroke in evolution")
→ Symptoms often on WAKING UP (overnight platelet aggregation)
→ NO sudden reperfusion → NO hemorrhagic transformation
- History of hypertension, hyperlipidemia, DM, smoking
- Carotid bruit, prior TIAs in same territory
- Preceded by crescendo TIAs (same vascular territory each time)
- MRI: pale/bland infarct, no blood
- Treatment: Antiplatelet (aspirin ± clopidogrel) + statin + BP control
🟠 EMBOLIC STROKE (CARDIOEMBOLIC = COMMONEST)
Cardiac stasis (AF, MI, dilated CMP, valvular disease)
→ LOW SHEAR → FIBRIN-RICH "RED THROMBUS" (platelet-poor, RBC-rich)
→ Loosely packed fibrin → EASY plasmin access → FAST LYSIS
→ Embolus travels → lodges distally (end-artery, NO collaterals)
→ Dense ischemia → blood-brain barrier SEVERELY DAMAGED
→ Clot lyses fast (spontaneous or tPA) → SUDDEN REPERFUSION
→ Damaged capillaries cannot tolerate → BLOOD EXTRAVASATES
→ HEMORRHAGIC TRANSFORMATION / HEMORRHAGIC INFARCT ✓
- AF (#1 cardioembolic cause — 5× stroke risk)
- Anterior MI → mural thrombus → embolism
- Mitral stenosis → LA appendage thrombus
- Prosthetic valve, infective endocarditis, atrial myxoma
- Sudden maximal-at-onset deficit (embolus hits fast)
- Multiple bilateral cortical strokes → always think embolic
- MRI: hemorrhagic infarct, cortical, wedge-shaped
- Treatment: Anticoagulation (NOT antiplatelet)
🟡 LACUNAR STROKE (Bonus — Small Vessel)
HTN + DM → lipohyalinosis of deep penetrating arteries
→ Lenticulostriate, thalamic, pontine arteries
→ Pure motor / pure sensory / ataxic-hemiparesis / clumsy-hand dysarthria
→ NO cortical signs (no aphasia, no hemianopia)
→ SIZE < 15 mm on MRI
→ Treatment: Antiplatelet
🔵 THE CORE COMPARISON TABLE
| Feature | Thrombotic | Cardioembolic | Lacunar |
|---|---|---|---|
| Vessel | Large artery (MCA, ICA) | Distal branches | Small deep arteries |
| Thrombus type | White (platelet-rich) | Red (fibrin-rich) | Lipohyalinosis |
| Onset | Stuttering / on waking | Sudden, maximal | Sudden |
| Preceded by TIA | Same territory | Different territory | Rare |
| Hemorrhagic Tx | ✗ | ✓ | ✗ |
| Imaging | Bland infarct | Hemorrhagic, cortical | <15mm, deep |
| Treatment | Antiplatelet + statin | Anticoagulate | Antiplatelet |
| #1 cause | HTN/DM/smoking | AF | HTN + DM |
⚡ WHY LYSIS SPEED DIFFERS — 3-LINE VERSION
THROMBOTIC → platelet-rich WHITE clot → dense cross-linked fibrin
→ Plasmin can't penetrate easily → SLOW lysis → no hemorrhage
EMBOLIC → fibrin-rich RED clot (low shear, cardiac) → loosely packed
→ Plasmin penetrates fast → FAST lysis → sudden reperfusion
→ Damaged BBB + end-artery territory → HEMORRHAGIC INFARCT
🔥 HIGH-YIELD ONE-LINERS (Exam Killers)
- AF + stroke → anticoagulate (warfarin/DOAC), NOT aspirin
- Cardioembolic stroke + tPA → increased risk of hemorrhagic transformation → still give tPA within window, but warn
- Marantic endocarditis (NBTE) = fibrin-platelet sterile vegetations on mitral/aortic → SLE, cancer → cerebral embolism
- Libman-Sacks = SLE → mitral valve (both surfaces) → embolic stroke
- Paradoxical embolus = DVT + PFO → venous clot crosses to arterial side → stroke in young patient
- AF stroke risk = paroxysmal AF = persistent AF (same risk — both need anticoagulation)
- Thrombotic stroke on waking = platelets most aggregable in early morning (platelet-activating factors peak)
- Hemorrhagic infarct ≠ hemorrhagic stroke — HI is reperfusion into dead tissue; HS is primary bleed
- tPA contraindicated if hemorrhagic transformation already visible on CT
- Cocaine/amphetamines → vasospasm → thrombotic stroke (common in young patients on Step 1)
🧠 EXAM VIGNETTE PATTERN RECOGNITION
| Vignette clue | Think |
|---|---|
| "Woke up with weakness" | Thrombotic |
| "Sudden maximal deficit while active" | Embolic |
| "Irregular rhythm on ECG" | Cardioembolic (AF) |
| "Recent anterior MI + new weakness" | Mural thrombus embolism |
| "Bilateral cortical strokes" | Embolic (proximal source) |
| "Young patient, DVT + stroke" | Paradoxical embolism via PFO |
| "Mitral stenosis + stroke" | LA appendage thrombus |
| "Atrial myxoma + stroke" | Embolism of tumor fragment |
| "SLE + stroke" | Libman-Sacks endocarditis |
| "Cancer + stroke" | Marantic endocarditis (NBTE) |
lenticulostraite vessel pic show me from circle of willis mca and mca branches at tthe beginning small vessels LSV supply the deeper parts of brain
lenticulostriate arteries MCA branches circle of Willis diagram
middle cerebral artery lenticulostriate vessels basal ganglia internal capsule supply
🧠 Image 1 — Schematic: MCA segments + LSA origin (coronal view)
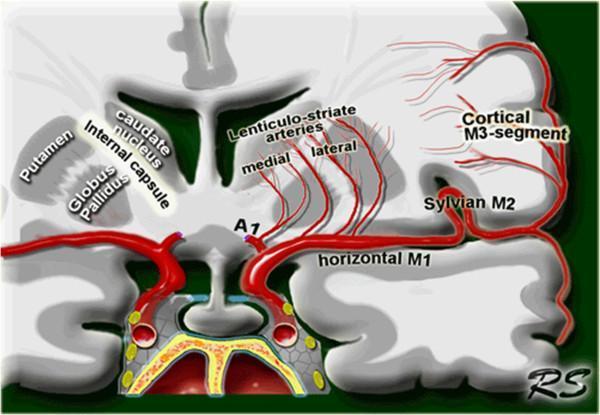
- Bottom = Circle of Willis / ICA → MCA stem (M1 = horizontal segment)
- A1 = ACA arising at the same level
- From M1, small vessels shoot straight up = medial + lateral lenticulostriate arteries (LSA)
- These supply → caudate, internal capsule, putamen, globus pallidus (the deep stuff)
- MCA then curves into the Sylvian fissure → M2 (Sylvian) → M3 (cortical) → cortex
🧠 Image 2 — Clean labeled coronal anatomy diagram
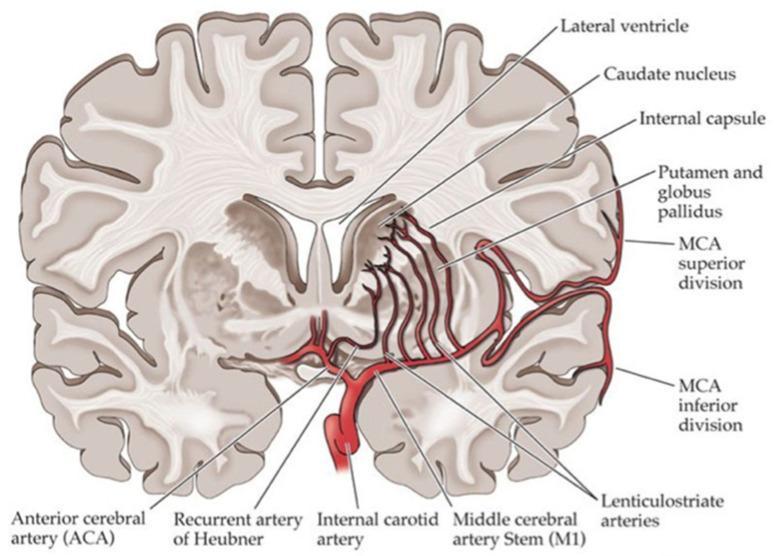
- ICA → bifurcates into ACA (A1) + MCA (M1)
- Recurrent artery of Heubner = from ACA → supplies head of caudate + anterior internal capsule
- Lenticulostriate arteries = from M1 → shoot up → supply putamen, globus pallidus, internal capsule, caudate body
- MCA superior + inferior divisions = cortical supply (lateral surface)
🧠 Image 3 — Real DSA angiogram showing LSA visibility depending on occlusion level
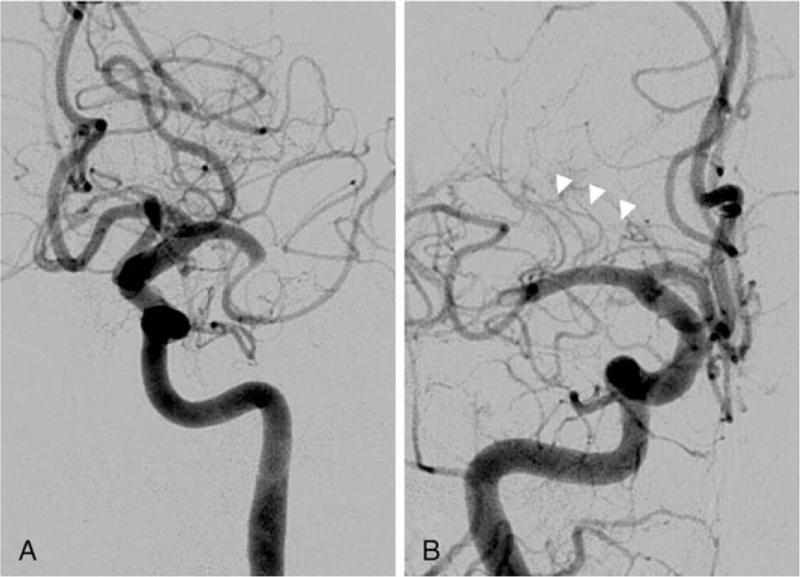
- Panel A = Proximal M1 occlusion → LSA origin is blocked → LSAs not visible → basal ganglia + internal capsule infarcted → dense hemiplegia
- Panel B = Distal M1 occlusion → LSAs still arise before the clot → LSAs spared (arrowheads) → basal ganglia preserved → mostly cortical deficit
⚡ Summary Arrow (copy to notes)
ICA → bifurcates at Circle of Willis
├─ ACA (A1) → Recurrent artery of Heubner → head of caudate + ant. internal capsule
└─ MCA (M1 = horizontal) → Lenticulostriate arteries (END ARTERIES, no collaterals)
↓ shoot straight up into deep brain
→ Putamen / Globus pallidus / Internal capsule / Caudate body
→ MCA continues → M2 (Sylvian fissure) → M3 (cortical branches) → lateral cortex
LSA = end arteries → HTN → lipohyalinosis → rupture/occlusion → LACUNAR STROKE
what is lucid interval in epidural hematoma
Lucid Interval in Epidural Hematoma
The Mechanism — Why It Happens
Head trauma (temporal region)
→ Temporal bone fracture → MIDDLE MENINGEAL ARTERY lacerated
→ Arterial bleed → blood accumulates in epidural space (between skull & dura)
→ Initial concussive LOC (brief, seconds–minutes) — from impact force
→ Blood volume still small → brain not yet compressed → patient WAKES UP
↓
★ LUCID INTERVAL (minutes to hours) ★
Patient is conscious, talking, seemingly fine
↓
→ Arterial bleeding CONTINUES (it's an artery — doesn't clot easily)
→ Hematoma expands → dura stripped from inner skull → ICP rises
→ Temporal lobe herniates medially → compresses CN III
→ Ipsilateral fixed, dilated pupil ("blown pupil") ← CN III compression
→ Contralateral hemiparesis (cerebral peduncle compressed)
→ Rapid LOC, coma → DEATH if not evacuated
Why the Lucid Interval is UNIQUE to EDH
| Feature | EDH | SDH | ICH |
|---|---|---|---|
| Vessel | Middle meningeal artery | Bridging veins | Parenchymal vessel |
| Bleed type | Arterial (high pressure) | Venous (slow) | Variable |
| Lucid interval | Classic (30–50% of cases) | Rare (most are drowsy from onset) | No |
| Onset | Rapid (minutes–hours) | Slow (hours–days) | Variable |
| CT shape | Biconvex / lenticular (lens-shaped) | Crescent-shaped | Irregular |
| Crosses sutures | NO (dura tightly attached) | Yes | N/A |
| Crosses midline | No | Yes (subdural space is continuous) | N/A |
CT Image — Acute EDH
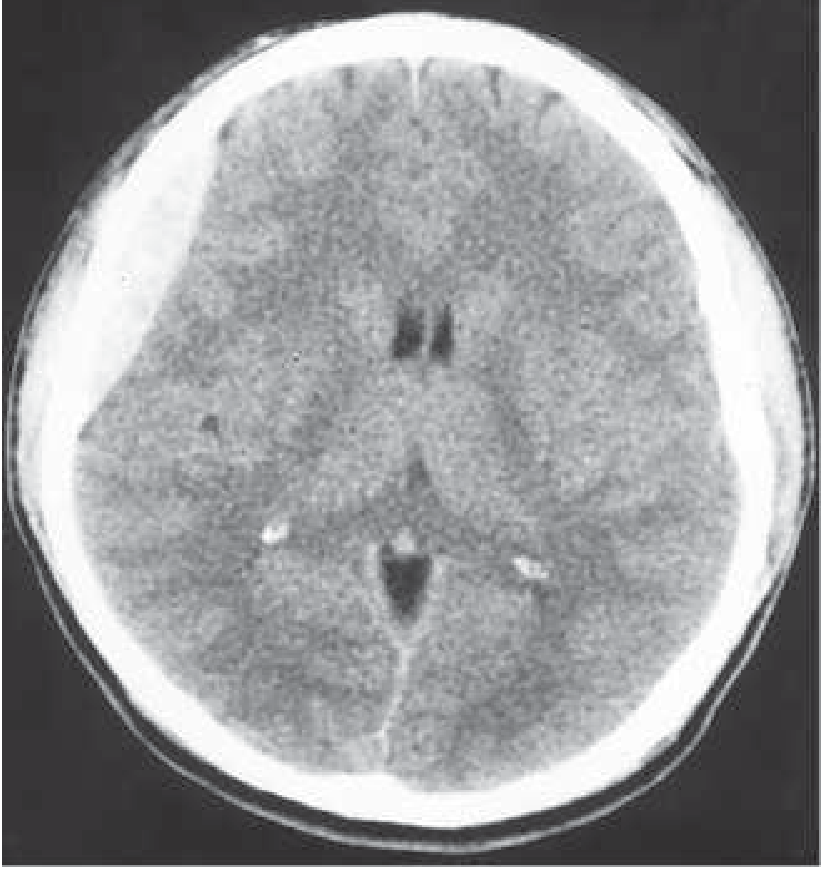
⚡ USMLE One-Liners (All Tested)
- Classic triad: head trauma → brief LOC → lucid interval → rapid deterioration
- Lucid interval present in only ~1/3 to 1/2 of EDH cases — absence does NOT rule it out
- Ipsilateral blown pupil = CN III compression by uncal herniation = surgical emergency
- "Talk and die" syndrome = patient was talking (lucid interval) → dies = classic EDH story
- Evacuate within 70 minutes — EDH cannot be watched; it's arterial, it keeps bleeding
- Burr hole = ipsilateral side of the dilated pupil (same side as hematoma)
- Temporal bone fracture is the key — pterion is thinnest part of skull → most vulnerable
- EDH does NOT cross suture lines (dura is tightly attached at sutures) — biconvex shape on CT
- SDH crosses suture lines (subdural space is open) — crescent shape on CT
⚡ Summary Arrow (copy to notes)
Temporal trauma → temporal bone Fx → Middle Meningeal Artery tear
→ Arterial bleed → small volume initially → patient wakes up = LUCID INTERVAL
→ Artery keeps bleeding → hematoma expands → ICP↑ → uncal herniation
→ CN III compressed → ipsilateral fixed dilated pupil
→ Cerebral peduncle compressed → contralateral hemiparesis
→ Rapid coma → death
Rx: Emergency craniotomy/burr hole within 70 min
CT: Biconvex hyperdense, does NOT cross sutures
— Harrison's Principles of Internal Medicine 22e; Schwartz's Principles of Surgery 11e
Hey, can you tell me what is Metachromatic Leukodystrophy? What do we have a deficiency of? Are enzyme sulfatase and sulfatides get accumulate, cannot be degraded, sulfatides or myelin which one cannot be degraded?
Metachromatic Leukodystrophy (MLD)
The Core Question: What accumulates and what can't be degraded?
NORMAL:
Myelin turnover → myelin breaks down → releases SULFATIDES (galactosylceramide sulfate)
→ Sulfatides enter lysosome
→ Arylsulfatase A (ARSA) cleaves the sulfate group
→ Sulfatide → Cerebroside (harmless, recycled)
IN MLD:
ARSA enzyme DEFICIENT (mutation in ARSA gene, autosomal recessive)
→ Sulfatides CANNOT be cleaved → accumulate in lysosomes
→ Build up inside: oligodendrocytes (CNS) + Schwann cells (PNS) + macrophages
→ Sulfatide accumulation → destabilizes myelin membrane
→ Myelin breaks down secondarily (demyelination)
→ White matter destroyed = LEUKODYSTROPHY
- Enzyme deficient = Arylsulfatase A (ARSA)
- What accumulates = SULFATIDES (the substrate)
- What cannot be degraded = SULFATIDES (not myelin — myelin is the victim, not the substrate)
- Myelin breaks down as a consequence of sulfatide toxicity, not because myelin itself can't be degraded
Why "Metachromatic"?
Sulfatides stain BROWN-ORANGE (not purple) with aniline dyes
= "metachromasia" = color change from the expected stain color
→ That's literally why the disease is called METAchromatic
MRI of MLD (2-year-old girl, T2-weighted)
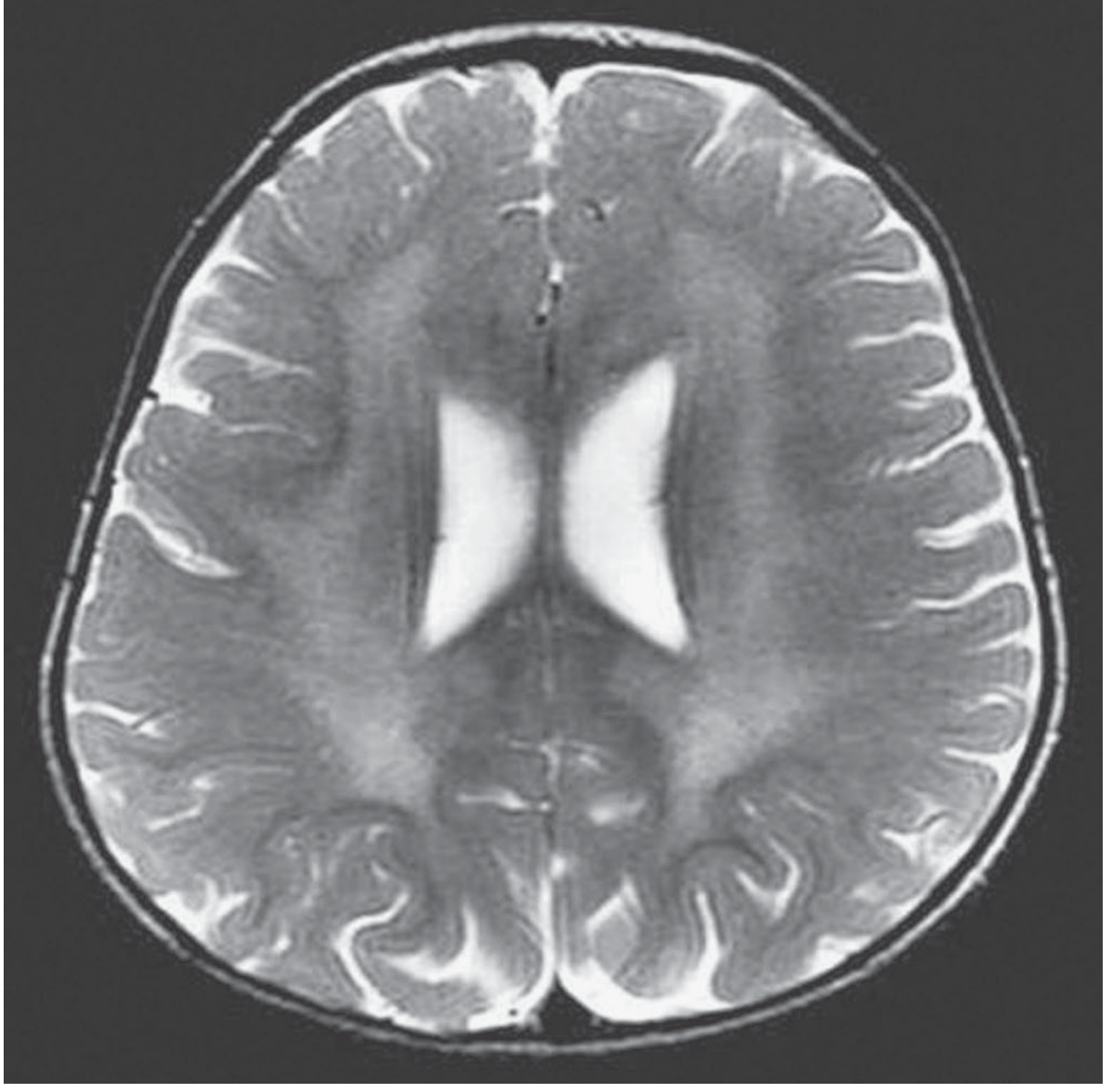
⚡ USMLE Master Summary
MLD = Lysosomal storage disease (sphingolipidosis / sulfatidosis)
↓
Deficient enzyme: ARYLSULFATASE A (ARSA)
↓
Substrate that accumulates: SULFATIDES (galactosylceramide-3-sulfate)
↓
Sulfatides build up in: oligodendrocytes (CNS) + Schwann cells (PNS)
↓
Myelin membrane destabilized → demyelination (secondary effect)
↓
White matter destroyed → leukodystrophy
↓
Clinical: progressive spastic paraparesis + peripheral neuropathy
(both CNS + PNS involved = distinguishing feature)
↓
AR inheritance | Most common: late infantile form (1–4 years onset)
↓
Labs: ↑ sulfatides in urine | ↓ ARSA in WBCs/fibroblasts | ↑ CSF protein
↓
Stain: metachromatic granules (brown-orange with aniline dyes, PAS+)
↓
MRI: bilateral symmetrical periventricular white matter T2 hyperintensity
Comparison with the close cousin — Krabbe Disease
| Feature | MLD | Krabbe |
|---|---|---|
| Enzyme deficient | Arylsulfatase A | Galactocerebrosidase (GALC) |
| Accumulates | Sulfatide | Galactocerebroside + Psychosine |
| Inheritance | AR | AR |
| Cells destroyed | Oligodendrocytes + Schwann cells | Oligodendrocytes (psychosine toxic) |
| Pathology hallmark | Metachromatic granules | Globoid cells (multinucleated macrophages) |
| Peripheral neuropathy | ✓ | ✓ |
— Goldman-Cecil Medicine; Adams & Victor's Principles of Neurology 12e; Robbins Pathologic Basis of Disease
Explain intranuclear ophthalmoplegia with a diagram, you know, a simplified diagram where I can see all these kinds of ophthalmoplegia because it is very, very confusing for me. And what are the high-yield ophthalmoplegias I have to remember in lens moves upward and downward. One side is that homonymous hemianopia, that, that, that. This is very, very confusing and I will get very stressed out when I see such questions. Can you make me a high yield table with simple, logical, and top level explanation so that I can remember and sort them for my SMV step exams?
internuclear ophthalmoplegia MLF diagram eye movement pathway pons midbrain
visual field defects optic pathway lesion diagram bitemporal hemianopia homonymous hemianopia quadrantanopia
Hey, I want you to explain what is internuclear ophthalmoplegia, and for me, I have the guess fear for all this kind of eye anopia, homonymous hemianopia, homonymous hemianopia, something like that, contralateral hemianopia, and all this kind of eye pictures. It's like if I'll confuse and put some wrong answer. I want some cheat sheet so that I can solve this very, very simply for USMLE step complex exams.
i do have
i have top up 250 cerdits