give me about diabetes in detail for Post graduate level
Reading File
Finding Sources
Finding Sources
Searching PubMed
"diabetes mellitus"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I have excellent comprehensive content from multiple authoritative textbooks. Now I'll write the full postgraduate-level response.
Diabetes Mellitus - Postgraduate Level Review
Sources: Robbins & Kumar Basic Pathology, Robbins Cotran & Kumar Pathologic Basis of Disease, Harrison's Principles of Internal Medicine 22E (2025), Tietz Textbook of Laboratory Medicine 7E, Katzung's Basic and Clinical Pharmacology 16E, Goldman-Cecil Medicine, Harrison's 22E
1. DEFINITION AND EPIDEMIOLOGY
Diabetes mellitus is a group of metabolic diseases characterized by chronic hyperglycemia resulting from defects in insulin secretion, insulin action, or both. The resulting dysregulation of carbohydrate, fat, and protein metabolism causes long-term damage, dysfunction, and failure of multiple organ systems, particularly the kidneys, eyes, nerves, heart, and blood vessels.
Globally, diabetes affects over 500 million adults. Type 2 diabetes (T2D) accounts for 90-95% of all cases. Type 1 diabetes (T1D) accounts for 5-10% of cases and represents the predominant form diagnosed in patients under 20 years of age.
2. CLASSIFICATION
The current classification (ADA, modified) recognizes four major categories:
Type 1 Diabetes (T1D)
- Immune-mediated (Type 1A): T-cell-mediated autoimmune destruction of pancreatic β-cells, leading to absolute insulin deficiency
- Idiopathic (Type 1B): Autoantibody-negative; rare
Type 2 Diabetes (T2D)
- Combination of peripheral insulin resistance and relative insulin secretory deficiency
- Eventually progresses to absolute insulin deficiency in many patients
Other Specific Types
| Category | Examples |
|---|---|
| Genetic defects in β-cell function | MODY (maturity-onset diabetes of the young) - HNF-1α, GCK, etc. |
| Genetic defects in insulin action | Severe insulin resistance syndromes, leprechaunism |
| Pancreatic disease ("Pancreaticogenic") | Chronic pancreatitis, cystic fibrosis, hemochromatosis, pancreatic carcinoma |
| Endocrinopathies | Cushing syndrome, acromegaly, pheochromocytoma, glucagonoma, hyperthyroidism |
| Drug/chemical-induced | Glucocorticoids, thiazides, protease inhibitors, β-adrenergic agonists, interferon-α |
| Genetic syndromes | Down syndrome, Klinefelter syndrome, Turner syndrome |
Gestational Diabetes Mellitus (GDM)
- Approximately 5% of pregnancies in the US are complicated by hyperglycemia
- Pregnancy is inherently a diabetogenic state (progesterone, human placental lactogen promote insulin resistance)
- Typically resolves post-delivery; risk of developing overt T2D is highest in the first 5 years postpartum
3. GLUCOSE HOMEOSTASIS AND INSULIN PHYSIOLOGY
Normal Insulin Secretion
Insulin is secreted by pancreatic β-cells in a biphasic pattern:
- First phase: Rapid spike within 1-3 minutes of glucose stimulus (exocytosis of preformed insulin granules)
- Second phase: Sustained release requiring new insulin synthesis
Key triggers: Glucose (primary), amino acids, GLP-1, GIP, acetylcholine. Glucose enters β-cells via GLUT-2, is phosphorylated by glucokinase (the "glucose sensor"), and ATP generated from metabolism closes K-ATP channels → membrane depolarization → Ca²⁺ influx → exocytosis.
Insulin Signaling
Insulin binds its receptor (a transmembrane receptor tyrosine kinase), triggering:
- IRS-1/PI3K/Akt pathway: Mediates GLUT-4 translocation to the cell surface (glucose uptake in muscle and fat), glycogen synthesis, lipogenesis, protein synthesis
- MAPK pathway: Cell growth and differentiation
- Net metabolic effects: ↑ glucose uptake by muscle/adipose, ↑ glycogenesis, ↓ hepatic gluconeogenesis, ↑ lipogenesis, ↓ lipolysis, ↑ protein synthesis
4. PATHOGENESIS
4A. Type 1 Diabetes
T1D results from cellular-mediated autoimmune destruction of insulin-secreting β-cells. The α-, δ-, and other islet cells are preserved. The islets show a chronic mononuclear cell infiltrate termed insulitis. An 80-90% reduction in β-cell volume is required before symptomatic diabetes appears. The autoimmune process begins months to years before clinical presentation, and the rate of islet destruction is typically more rapid in children than adults.
Genetic susceptibility:
- The HLA region on chromosome 6p21 confers ~50% of genetic risk
- HLA-DR3 and HLA-DR4 (or DR3/DR4 heterozygosity) are the highest risk haplotypes
- HLA-DQ alleles in linkage disequilibrium with DR alleles play a key mechanistic role - protective and predisposing alleles affect thymic presentation of insulin peptides
- Polymorphisms in the insulin gene (VNTR in the promoter region) affect thymic expression of insulin and contribute to susceptibility - low expression → impaired central tolerance to insulin
- Other non-HLA genes: PTPN22, CTLA4, IL-2RA (CD25)
Autoimmune markers (key islet autoantibodies):
| Autoantibody | Features |
|---|---|
| ICA (Islet cell cytoplasmic antibodies) | Present in 75-85% of newly diagnosed T1D; detected in 0.5% of normals |
| IAA (Insulin autoantibodies) | Present in ~80-90% of children developing T1D before age 5; <40% in those diagnosed after age 12 |
| GADA (Anti-GAD65) | Detected up to 10 years before clinical onset; present in ~60% of newly diagnosed; key marker for LADA |
| Anti-IA-2 (Tyrosine phosphatase) | ICA512; present in ~50-70% |
| Anti-ZnT8 (Zinc transporter 8) | Present in ~60-80% of new-onset T1D |
Mechanisms of β-cell destruction:
- CD4+ and CD8+ T cells are the primary effectors
- CD8+ cytotoxic T cells directly kill β-cells via perforin/granzyme and Fas-FasL interactions
- Inflammatory cytokines (IL-1β, TNF-α, IFN-γ) from macrophages cause local inflammation and β-cell apoptosis
- Loss of regulatory T cell (Treg) suppression allows uncontrolled autoimmunity
Environmental factors:
- Viral triggers (Coxsackie B virus, enterovirus) - molecular mimicry hypothesis
- Gut microbiome dysbiosis
- The "hygiene hypothesis" - reduced early microbial exposure
- Early cow's milk exposure has been proposed but recent trials (TRIGR study with 2,159 at-risk neonates, 11.5 years median follow-up) showed no effect
Staging (Current ADA Framework):
- Stage 1: ≥2 autoantibodies, normoglycemia, asymptomatic
- Stage 2: ≥2 autoantibodies, dysglycemia (impaired fasting glucose or IGT), asymptomatic
- Stage 3: Clinical diabetes (overt hyperglycemia, symptoms)
Teplizumab (anti-CD3 monoclonal antibody) - FDA approved for Stage 2 T1D in patients ≥8 years. 14-day IV infusion delays progression to Stage 3 by a median of ~25 months. Delays progression by modulating pathogenic T-cell responses. - Katzung's Pharmacology, p.1174
4B. Type 2 Diabetes
T2D is an extremely heterogeneous disease with at least two major pathologic defects:
1. Insulin Resistance
Defined as a decreased biological response to normal concentrations of circulating insulin. It is found in obese non-diabetic individuals and in all patients with T2D, and may precede clinical diabetes by up to 20 years.
Mechanisms:
- Post-receptor defects: Impaired IRS-1/PI3K/Akt signaling
- Systemic inflammation: Elevated IL-6 and TNF-α from adipose tissue (particularly visceral fat) impair insulin signaling in liver and muscle
- Lipotoxicity: Elevated free fatty acids impair insulin signaling and promote β-cell apoptosis
- Ectopic fat deposition in liver (NAFLD/MASLD) and muscle causes local insulin resistance
- Mitochondrial dysfunction in skeletal muscle
2. Progressive β-cell failure
- Initial compensation: β-cells hypersecrete insulin to maintain normoglycemia against insulin resistance
- Selective glucose unresponsiveness (glucotoxicity): Chronic hyperglycemia impairs glucose-stimulated insulin release; restoring euglycemia rapidly resolves this
- Loss of first-phase insulin secretion is an early and specific defect
- β-cell dedifferentiation (not just apoptosis) is a proposed mechanism - mature β-cells revert to progenitor-like states
- Islet amyloid polypeptide (IAPP/amylin) deposition contributes to β-cell dysfunction and loss
- Number of β-cells is reduced by 20-65% in T2D at autopsy
Other contributors:
- Increased hepatic glucose production (gluconeogenesis and glycogenolysis)
- α-cell dysfunction: Inappropriately elevated glucagon (bihormonal hypothesis)
- Incretin defect: Impaired GLP-1 response to meals
- Renal glucose reabsorption: Upregulated SGLT2 expression
4C. MODY (Maturity-Onset Diabetes of the Young)
Monogenic diabetes caused by single-gene mutations in genes encoding proteins involved in β-cell development and function. Autosomal dominant inheritance; typically presents before age 25. Often misclassified as T1D or T2D.
| Type | Gene | Key Features |
|---|---|---|
| MODY 1 | HNF-4α | Progressive β-cell failure; sensitive to sulfonylureas |
| MODY 2 | Glucokinase (GCK) | Mild stable fasting hyperglycemia; rarely requires treatment |
| MODY 3 | HNF-1α | Most common; progressive; sensitive to sulfonylureas; low renal threshold for glucose (glycosuria at lower glucose levels) |
| MODY 5 | HNF-1β | Associated with renal developmental anomalies |
5. DIAGNOSTIC CRITERIA (ADA)
| Test | Diabetes | Prediabetes |
|---|---|---|
| Fasting plasma glucose | ≥ 126 mg/dL (7.0 mmol/L) | 100-125 mg/dL (IFG) |
| 2-hour plasma glucose (75g OGTT) | ≥ 200 mg/dL (11.1 mmol/L) | 140-199 mg/dL (IGT) |
| HbA1c | ≥ 6.5% (48 mmol/mol) | 5.7-6.4% |
| Random plasma glucose + symptoms | ≥ 200 mg/dL | — |
- In the absence of symptoms, two abnormal tests from the same or different samples are required to confirm diagnosis
- HbA1c is not appropriate for diagnosis in hemolytic anemias, hemoglobinopathies, pregnancy, or end-stage renal disease
6. CLINICAL FEATURES
T1D
- Classic triad: Polyuria, polydipsia, polyphagia with weight loss
- Onset often acute/dramatic, may present with DKA
- Age: Commonly <30 years; bimodal distribution (childhood/early adulthood + 40-50s)
- "Honeymoon phase" (first 1-2 years): Residual β-cells may allow reduced insulin requirement
T2D
- Often asymptomatic at diagnosis (picked up on screening)
- May present with recurrent infections (UTI, candidiasis, skin), blurred vision, fatigue, or complications
- Associated with obesity, metabolic syndrome, acanthosis nigricans
- Many require insulin eventually due to progressive β-cell failure
7. ACUTE COMPLICATIONS
7A. Diabetic Ketoacidosis (DKA)
Biochemical triad:
- Hyperglycemia (or prior history of diabetes) - "D"
- Urinary ketones ≥2+ or blood ketones ≥3.0 mmol/L - "K"
- Arterial/venous pH <7.3 - "A"
Pathophysiology: Absolute or relative insulin deficiency + counter-regulatory hormone excess (glucagon, cortisol, catecholamines, GH) →
- Unrestricted lipolysis → free fatty acids → hepatic ketogenesis (acetoacetate, β-hydroxybutyrate, acetone)
- Hepatic gluconeogenesis and glycogenolysis → hyperglycemia
- Osmotic diuresis → dehydration, electrolyte loss (K⁺, Na⁺, PO₄³⁻, Mg²⁺)
- Anion gap metabolic acidosis
Precipitants: Infection (most common), insulin omission, new-onset T1D, acute coronary syndrome, trauma, drugs (cocaine, SGLT2 inhibitors in euglycemic DKA)
Diagnosis: ABG, serum/urine ketones, electrolytes (calculate anion gap), BUN/Cr, ECG (K⁺ effect on cardiac conduction)
Management:
- Fluid resuscitation: 0.9% NaCl 15-20 mL/kg/hr initially; switch to 0.45% NaCl once Na normalized
- Insulin: Regular insulin IV infusion 0.1 U/kg/hr (after K⁺ ≥3.3 mEq/L); reduce to 0.02-0.05 U/kg/hr once glucose <200 mg/dL; add dextrose to IV fluid
- Potassium replacement: Critical - replace even if K⁺ is "normal" (total body depletion always present)
- Phosphate: Replace if <1.0 mg/dL or hemolytic anemia, cardiac dysfunction
- Bicarbonate: Only if pH <6.9
Resolution criteria: Glucose <200 mg/dL, anion gap ≤12, bicarbonate ≥15 mEq/L, pH >7.3
7B. Hyperosmolar Hyperglycemic State (HHS)
- Typically T2D, older patients
- Glucose often >600 mg/dL; serum osmolality >320 mOsm/kg
- Minimal or absent ketoacidosis (residual insulin prevents lipolysis)
- Profound dehydration (8-10 L deficit)
- Altered consciousness; mortality 10-20% (vs 1-5% for DKA)
- Treatment: Aggressive fluid replacement (0.45-0.9% NaCl), lower-dose insulin, careful monitoring
8. CHRONIC COMPLICATIONS
The unifying mechanism: Chronic hyperglycemia → microvascular and macrovascular damage via multiple pathways.
Molecular Mechanisms of Hyperglycemia-Induced Damage:
- Polyol pathway: Aldose reductase converts glucose to sorbitol (osmotic damage, oxidative stress) - relevant in retina, lens, peripheral nerves
- Advanced glycation end-products (AGEs): Non-enzymatic glycation of proteins/lipids → cross-linking, receptor activation (RAGE) → inflammation, vascular damage
- Protein kinase C (PKC) activation: DAG accumulation → PKC → VEGF overexpression (retinal neovascularization), TGF-β (glomerulosclerosis), NF-κB
- Hexosamine pathway: O-GlcNAc modification of transcription factors → impairs insulin signaling
- Oxidative stress: Mitochondrial superoxide overproduction is the common upstream trigger for all four pathways
8A. Diabetic Nephropathy (Diabetic Kidney Disease)
Leading cause of end-stage renal disease (ESRD) in the developed world.
Pathology (Robbins):
- Glomerular basement membrane (GBM) thickening: Detected by electron microscopy within a few years; precedes functional decline
- Diffuse mesangial sclerosis: Mesangial matrix expansion + cell proliferation; found in most patients with >10 years of disease
- Nodular glomerulosclerosis (Kimmelstiel-Wilson lesion): Ball-like deposits of laminated matrix in the mesangial periphery; occurs in 15-30% of long-term diabetics; virtually pathognomonic for diabetes - Robbins & Kumar Basic Pathology
- Hyaline arteriolosclerosis: Affects BOTH afferent AND efferent arterioles - involvement of efferent arteriole is virtually unique to diabetes
- Pyelonephritis: More severe in diabetics; necrotizing papillary necrosis more prevalent
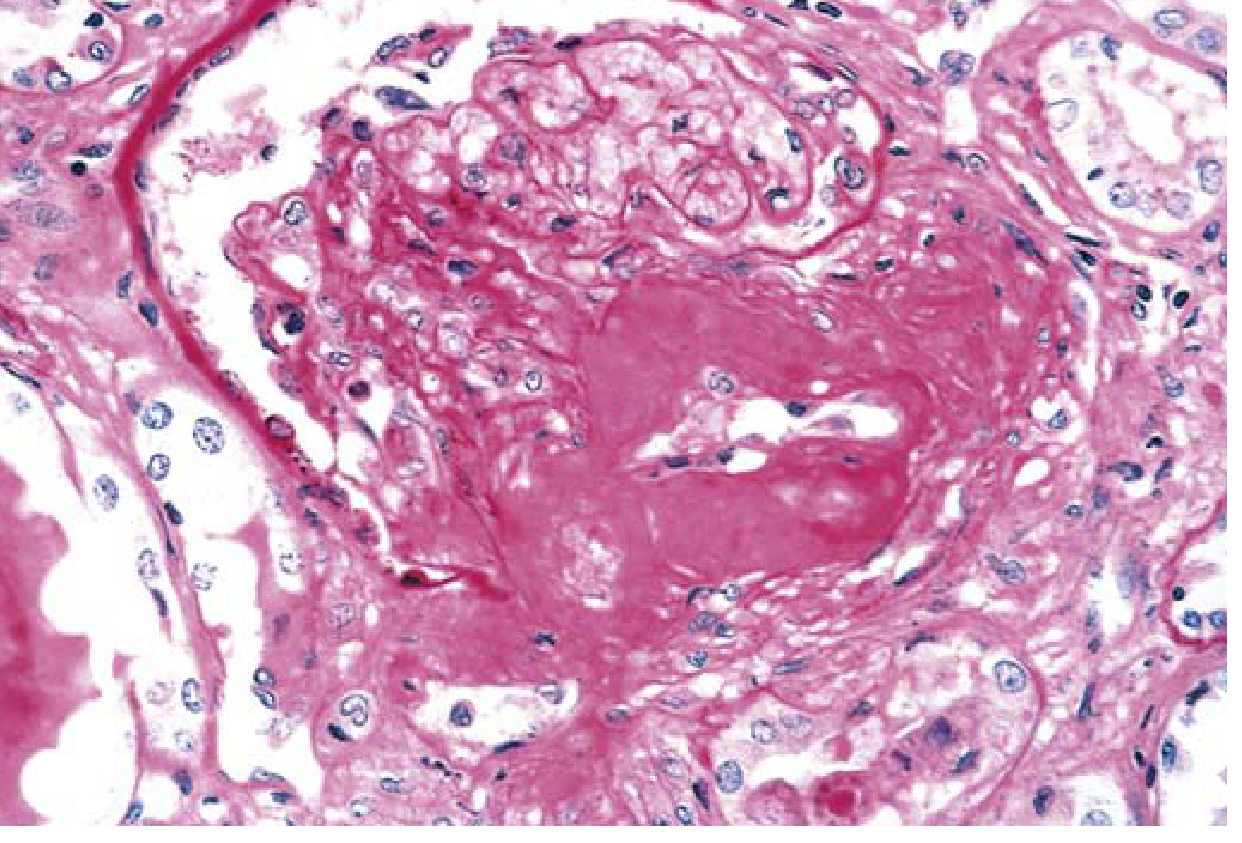
Severe renal hyaline arteriolosclerosis in diabetic nephropathy (PAS stain) - Robbins & Kumar Basic Pathology
Clinical stages (Mogensen classification for T1D):
- Renal hypertrophy, hyperfiltration (GFR elevated)
- Silent phase (microstructural changes, normal AER)
- Incipient nephropathy: Microalbuminuria 30-300 mg/24h (albuminuria 3-30 mg/mmol Cr)
- Overt nephropathy: Macroalbuminuria >300 mg/24h, declining GFR
- ESRD: GFR <15 mL/min
Screening: Annual urine albumin-to-creatinine ratio (UACR) from T1D diagnosis +5 years; from T2D diagnosis
Management: RAAS blockade (ACEi/ARB), tight glycemic control, SGLT2 inhibitors (renoprotective - CREDENCE, DAPA-CKD trials)
8B. Diabetic Retinopathy
Leading cause of new blindness in working-age adults.
Classification:
| Stage | Features |
|---|---|
| Background (Non-proliferative, NPDR) | Microaneurysms (earliest), dot/blot hemorrhages, hard exudates (lipid leakage), soft exudates (cotton-wool spots = nerve fiber infarcts), venous beading |
| Pre-proliferative NPDR | Venous beading, intraretinal microvascular abnormalities (IRMA), multiple cotton-wool spots |
| Proliferative (PDR) | New vessel formation (NVD - new vessels at disc; NVE - elsewhere), vitreous hemorrhage, tractional retinal detachment |
| Diabetic Macular Edema (DME) | Thickening of/near macula; major cause of vision loss in T2D |
Pathogenesis: Loss of pericytes (earliest change) → increased capillary permeability → microaneurysm formation → ischemia → VEGF overexpression → neovascularization
Management: Tight glycemic control (DCCT trial proved benefit), tight BP control, anti-VEGF agents (bevacizumab, ranibizumab, aflibercept) for PDR/DME, laser photocoagulation, vitrectomy
8C. Diabetic Neuropathy
Most common complication; affects ~50% of patients over time.
Types:
- Distal symmetric polyneuropathy (DSPN): Most common; "stocking-glove" sensory loss; starts with small fiber dysfunction (pain, temperature) then large fiber (vibration, proprioception, reflexes) → risk of painless trauma, Charcot's neuroarthropathy
- Autonomic neuropathy: Gastroparesis, orthostatic hypotension, erectile dysfunction, neurogenic bladder, diabetic diarrhea, gustatory sweating, impaired hypoglycemia awareness
- Radiculoplexus neuropathy (Bruns-Garland syndrome / diabetic amyotrophy): Severe asymmetric proximal leg pain, motor weakness; thought to be ischemic/inflammatory
- Cranial nerve palsies: Most commonly CN III with pupil sparing (ischemic, unlike compressive lesion)
- Mononeuropathy multiplex
Management: Tight glycemic control (primary prevention), pregabalin/gabapentin, duloxetine, TCAs (amitriptyline) for neuropathic pain; foot care and monitoring
8D. Macrovascular Complications
Atherosclerosis is accelerated and diffuse. Diabetics have:
- 2-4x increased risk of coronary artery disease (CAD) and myocardial infarction
- 2-4x increased risk of stroke
- Peripheral artery disease (PAD) - critical limb ischemia, diabetic foot
Diabetic cardiomyopathy: Heart failure even without CAD or hypertension - due to myocardial fibrosis, impaired Ca²⁺ handling, accumulation of AGEs
8E. Diabetic Foot
Results from the triad of: peripheral neuropathy + peripheral vascular disease + impaired immunity
- Neuropathic ulcers: Over pressure points, painless, good pulses
- Ischemic ulcers: Painful, poor pulses
- Wagner classification (Grade 0-5) used for staging
- MRSA, Pseudomonas, anaerobes commonly involved in deep infections
9. PHARMACOLOGICAL MANAGEMENT
9A. Insulin Therapy
Insulin preparations by duration:
| Type | Examples | Onset | Peak | Duration |
|---|---|---|---|---|
| Rapid-acting analogs | Lispro (Humalog), Aspart (NovoLog/Fiasp), Glulisine (Apidra) | 5-15 min | 1-1.5 h | ~4 h |
| Short-acting (Regular) | Humulin R, Novolin R | 30 min | 2 h | 5-7 h |
| Intermediate-acting | NPH (isophane) | 1-2 h | 4-8 h | 10-18 h |
| Long-acting analogs | Glargine (Lantus/Basaglar), Detemir (Levemir) | 1-2 h | Peakless/flat | 20-24 h |
| Ultra-long-acting | Degludec (Tresiba) | Slow | Peakless | >42 h |
| Concentrated | U500 Regular, U300 Glargine | — | — | Extended vs. U100 |
Key points from Katzung:
- Rapid-acting analogs (lispro, aspart, glulisine): Inject immediately before meals; optimal A1c reduction with less hypoglycemia than regular insulin; ~5% variability vs. 25% for regular insulin
- Fiasp (ultra-rapid aspart with niacinamide): Onset ~10 min faster than standard aspart
- U500 Regular insulin: Has a time-action profile similar to NPH (not regular) - important clinical distinction
- Intravenous insulin (DKA, perioperative): Regular insulin only
9B. Non-Insulin Agents
| Class | Drug Examples | Mechanism | Key Notes |
|---|---|---|---|
| Biguanides | Metformin | Activates AMPK → ↓ hepatic glucose production; improves insulin sensitivity | First-line; weight neutral/slight loss; GI side effects; rare lactic acidosis; hold for contrast/surgery; UKPDS cardiovascular benefit |
| SGLT2 inhibitors | Empagliflozin, Dapagliflozin, Canagliflozin | Block glucose reabsorption in proximal tubule → glucosuria | CV and renal protective benefits (EMPA-REG, DECLARE, CREDENCE); HF benefit; risk of UTI, DKA (euglycemic) |
| GLP-1 receptor agonists | Semaglutide, Liraglutide, Dulaglutide, Exenatide | Stimulate insulin, suppress glucagon, delay gastric emptying, promote satiety | CV benefit in high-risk T2D (LEADER, SUSTAIN-6); weight loss; weekly/daily SC or oral (semaglutide); avoid in MEN2/medullary thyroid ca |
| DPP-4 inhibitors | Sitagliptin, Saxagliptin, Alogliptin, Linagliptin | Inhibit DPP-4 → ↑ endogenous GLP-1/GIP | Weight neutral; well tolerated; saxagliptin: ↑ HF hospitalization (SAVOR-TIMI) |
| Sulfonylureas | Glibenclamide, Glipizide, Glimepiride, Gliclazide | Close K-ATP channels → insulin secretion (glucose-independent) | Risk of hypoglycemia and weight gain; prefer 2nd generation |
| Thiazolidinediones (TZDs) | Pioglitazone, Rosiglitazone | PPARγ agonists → improve insulin sensitivity | Fluid retention, CHF, fractures, bladder cancer risk (pioglitazone) |
| Alpha-glucosidase inhibitors | Acarbose, Miglitol | Delay carbohydrate absorption in gut | GI side effects; useful for postprandial hyperglycemia |
| Meglitinides | Repaglinide, Nateglinide | Close K-ATP channels (different binding site, rapid, short duration) | Taken with meals; flexible dosing |
| Dual GIP/GLP-1 agonist | Tirzepatide (Mounjaro) | Agonizes both GLP-1 and GIP receptors | Approved 2022 for T2D; superior A1c reduction and weight loss vs. GLP-1 alone; FDA approved for obesity 2023 |
10. GLYCEMIC TARGETS
| Parameter | General Target | Comment |
|---|---|---|
| HbA1c | <7.0% | <6.5% if feasible without hypoglycemia |
| Fasting/preprandial BG | 80-130 mg/dL | |
| 2-hr postprandial BG | <180 mg/dL | |
| BP | <130/80 mmHg | ACEi/ARB preferred if albuminuria |
| LDL-C | <70 mg/dL (high risk) | Statin therapy for all >40 yrs or with ASCVD |
Targets are individualized based on age, comorbidities, hypoglycemia risk, patient preference, and life expectancy.
11. MONITORING
- HbA1c: Every 3 months if not at goal; every 6 months if at goal. Reflects average glucose over ~3 months (RBC lifespan). Falsely low in hemolytic anemia, hemoglobinopathies, blood loss; falsely high in iron deficiency.
- Self-monitoring of blood glucose (SMBG): Frequency depends on treatment regimen
- Continuous glucose monitoring (CGM): Standard of care in T1D; increasingly used in T2D; metrics include time-in-range (TIR), GMI, CV%
- Annual screening: UACR, eGFR, fundoscopy, foot exam, lipids, BP, TSH (T1D)
12. SPECIAL SITUATIONS
Diabetes in Pregnancy
- Pre-gestational diabetes: Risk of congenital malformations (cardiac, neural tube) proportional to first-trimester HbA1c → target HbA1c 6-6.5%
- Macrosomia: Due to maternal hyperglycemia → fetal hyperinsulinism → anabolic growth
- Only insulin is approved for use throughout pregnancy (oral agents are used in some centers but lack definitive safety data)
- GDM screening: 24-28 weeks with 75g OGTT
LADA (Latent Autoimmune Diabetes in Adults)
- Up to 10-15% of "type 2" northern European patients actually have LADA
- GADA positive, slower β-cell destruction than T1D, presents >30 years
- Treatment: Same as T2D initially, but requires insulin sooner
13. SUMMARY TABLE: T1D vs T2D
| Feature | T1D | T2D |
|---|---|---|
| Prevalence | 5-10% | 90-95% |
| Age of onset | Typically <30 years | Typically >40 years (but rising in youth) |
| Body habitus | Normal/lean | Usually overweight/obese |
| Insulin | Absolute deficiency | Relative deficiency + resistance |
| Autoimmunity | Yes (autoantibodies) | No |
| HLA association | Yes (DR3/DR4) | No |
| Ketosis | Common | Rare (unless severe stress) |
| C-peptide | Low/undetectable | Normal/elevated early; low late |
| Genetics | Polygenic (HLA dominant) | Highly polygenic + environmental |
| Treatment | Always requires insulin | Lifestyle → oral agents → insulin |
Key References:
- Robbins & Kumar Basic Pathology, Chapter 18 (Endocrine Pancreas, pp. 744-757)
- Tietz Textbook of Laboratory Medicine, 7E, Chapter 47 (Carbohydrates, pp. 2294-2360)
- Katzung's Basic and Clinical Pharmacology, 16E, Chapter 41 (pp. 1173-1200)
- Goldman-Cecil Medicine, Chapter 210 (Acute Hyperglycemic States)
- Harrison's Principles of Internal Medicine, 22E (2025)
This is a shared conversation. Sign in to Orris to start your own chat.