Multiple hereditary exotosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"multiple hereditary exostoses"[MeSH Terms] OR "hereditary multiple exostoses"
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Here is the full answer:
Multiple Hereditary Exostoses (MHE)
Multiple hereditary exostoses (MHE), also called hereditary multiple exostoses or diaphyseal aclasis, is an autosomal dominant skeletal dysplasia characterized by the formation of multiple osteochondromas at the metaphyses of long bones.
Genetics and Pathogenesis
MHE is caused by germline mutations in one of two tumor suppressor genes:
- EXT1 - chromosome 8q24.11-q24.13
- EXT2 - chromosome 11p11-12
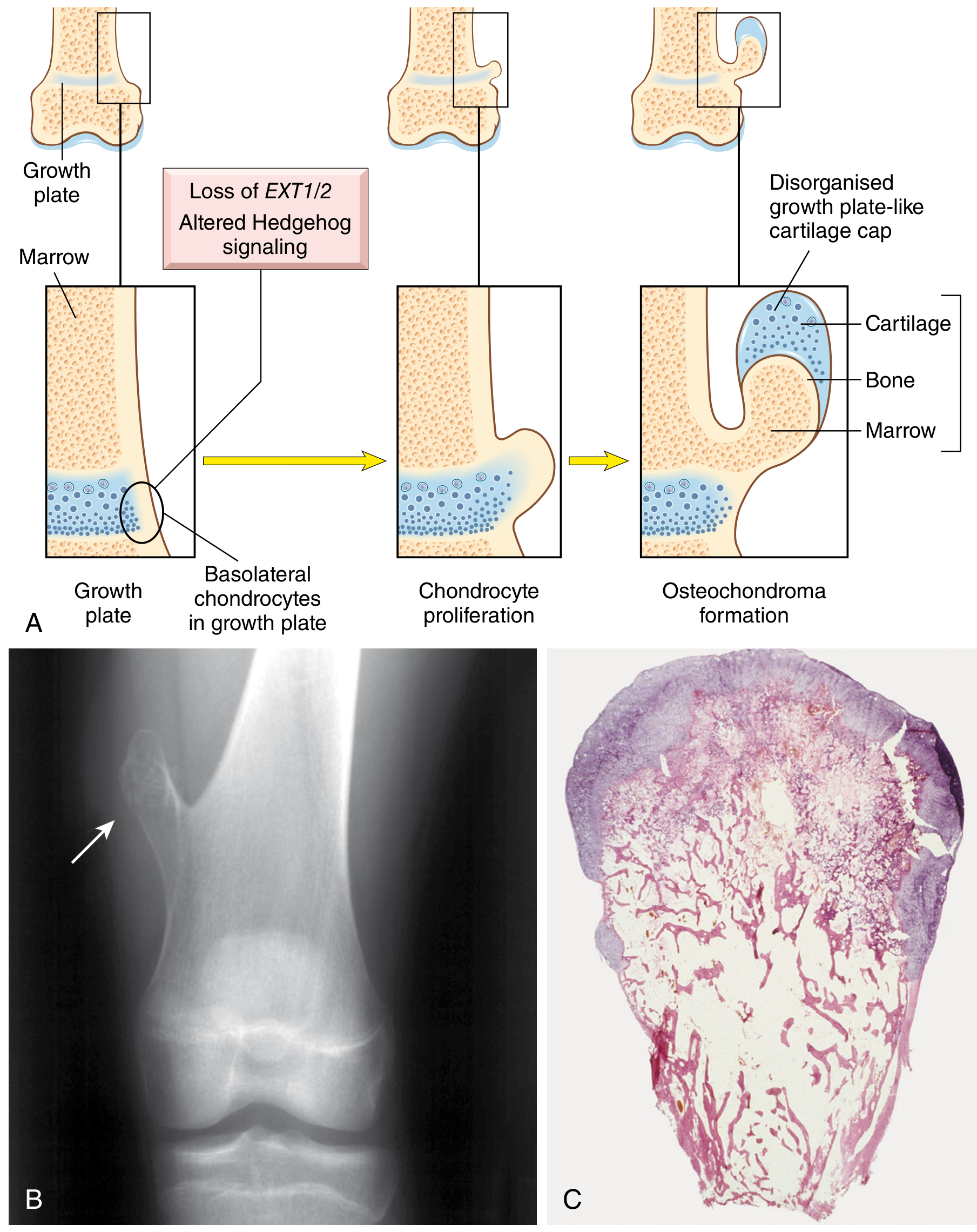
Both genes encode glycosyltransferases involved in heparan sulfate biosynthesis. Loss-of-function mutations (typically producing a truncated protein) result in reduced heparan sulfate, which disrupts the polarity and proliferation of chondrocyte progenitors in the growth plate. This secondarily perturbs signaling through the Hedgehog, BMP, FGFR3, and Wnt pathways, leading to lateral outgrowth of chondrocytes from the growth plate - the osteochondroma.
Two-hit model: The germline mutation is followed by somatic loss of the remaining wild-type allele in growth plate chondrocytes (loss of heterozygosity), consistent with a tumor suppressor mechanism.
EXT1 mutations are associated with a greater burden of disease and a higher risk of malignant transformation.
Epidemiology
- MHE occurs about 5-10% as frequently as solitary osteochondroma
- More common in males
- Autosomal dominant with variable (incomplete) penetrance
- Becomes apparent during childhood (vs. solitary osteochondromas, which present in late adolescence/early adulthood)
Pathology / Morphology
- Osteochondromas in MHE are often sessile and large (vs. pedunculated in solitary lesions)
- The bony stalk's cortex is continuous with the cortex of the host bone, and the medullary cavities communicate
- The surface cap is composed of disorganized hyaline cartilage (resembling a growth plate) covered by perichondrium
- Cap thickness: normally a few mm in adults, up to 2 cm in children
- Osteochondromas in MHE may be more cellular and display more atypia than the sporadic variety
- Endochondral ossification forms bone in the inner stalk
Clinical Features
| Feature | Details |
|---|---|
| Sites | Metaphyses of long bones - distal femur, proximal tibia, proximal humerus most common; also pelvis, scapula, ribs |
| Presentation | Multiple palpable masses, often discovered incidentally; may be painful if impinging on nerves or if stalk fractures |
| Skeletal deformity | Abnormal tubulation producing broad/blunt metaphyses; bowing of the radius, shortening of the ulna, producing ulnar deviation of the hand |
| Growth disturbance | Underlying bones may be bowed and shortened due to epiphyseal growth disturbance |
| Growth cessation | Exostoses stop growing at skeletal maturity (growth plate closure) |
Complications
1. Malignant Transformation (Chondrosarcoma)
- ~10% of MHE patients develop secondary chondrosarcoma (Miller's/Robbins)
- Some large referral-center series quote ~5%; the true community-level incidence is likely lower due to ascertainment bias
- Red flags for malignant transformation:
- Growth of a previously stable lesion in an adult
- New onset pain
- Cartilage cap thickness >2 cm on CT or MRI
- Evaluation: CT or MRI is the modality of choice for assessing cap thickness
2. Mechanical Complications
- Neurovascular compression (neuropathy, false aneurysms of major lower limb vessels)
- Overlying bursitis (bursa forms over tumor, may contain osteocartilaginous loose bodies)
- Fracture of the stalk
- Joint impingement
3. Skeletal Deformity
- Forearm deformity (radial bowing, ulnar shortening) is a major source of morbidity
- Limb length discrepancy
Imaging
- Plain radiographs: Usually sufficient for diagnosis - pedunculated or sessile bony lesion with the intramedullary canal continuous with that of the host bone; oriented away from the nearest physis; calcifications within the cap may be visible
- CT / MRI: Required to evaluate cartilage cap thickness and assess for malignant transformation; MRI best delineates soft-tissue extent
Treatment
| Indication | Approach |
|---|---|
| Asymptomatic lesions | Observation |
| Symptomatic (pain, nerve/vessel compression, cosmetic) | Surgical resection (en bloc excision, including the entire cartilage cap - recurrence is caused by incomplete cap removal) |
| Malignant transformation | Wide surgical resection (as for chondrosarcoma; chemotherapy has not shown survival benefit for chondrosarcoma) |
| Skeletal deformity | Osteotomies to correct bowing/angulation |
| Forearm deformity | Ulnar lengthening procedures (a 2026 systematic review [PMID 42116060] evaluated outcomes of ulnar lengthening for forearm deformities in MHE) |
MHE vs. Solitary Osteochondroma - Key Differences
| Feature | Solitary | MHE |
|---|---|---|
| Inheritance | Sporadic | Autosomal dominant |
| Genetics | Reduced EXT1/2 expression | Germline EXT1 or EXT2 mutation |
| Onset | Late adolescence/early adulthood | Childhood |
| Morphology | Often pedunculated | Often sessile, larger |
| Malignant risk | ~1% | ~5-10% |
| Skeletal deformity | Rare | Common (forearm, knee) |
Association Summary
- Syndrome associated with chondrosarcoma (secondary malignancy)
- EXT1 mutation = worse phenotype and higher malignancy risk
- MHE is the syndrome most commonly associated with secondary chondrosarcoma (among all bone tumor syndromes)
Sources: Robbins & Cotran Pathologic Basis of Disease, p. 1097; Miller's Review of Orthopaedics 9th ed., p. 822, 841; Campbell's Operative Orthopaedics 15th ed. 2026, p. 1163-1164
This is a shared conversation. Sign in to Orris to start your own chat.