Types of amyloidosis
Reading File
Finding Sources
Searching PubMed
amyloidosis classification types
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive content from Robbins' pathology textbook including the classification table and pathogenesis diagram. Let me compile the full answer.
Types of Amyloidosis
Amyloidosis is a group of disorders defined by the extracellular deposition of misfolded proteins in a beta-pleated sheet configuration. Over 30 amyloidogenic proteins are known. Current classification is based on the biochemical nature of the fibril protein, not clinical presentation alone.
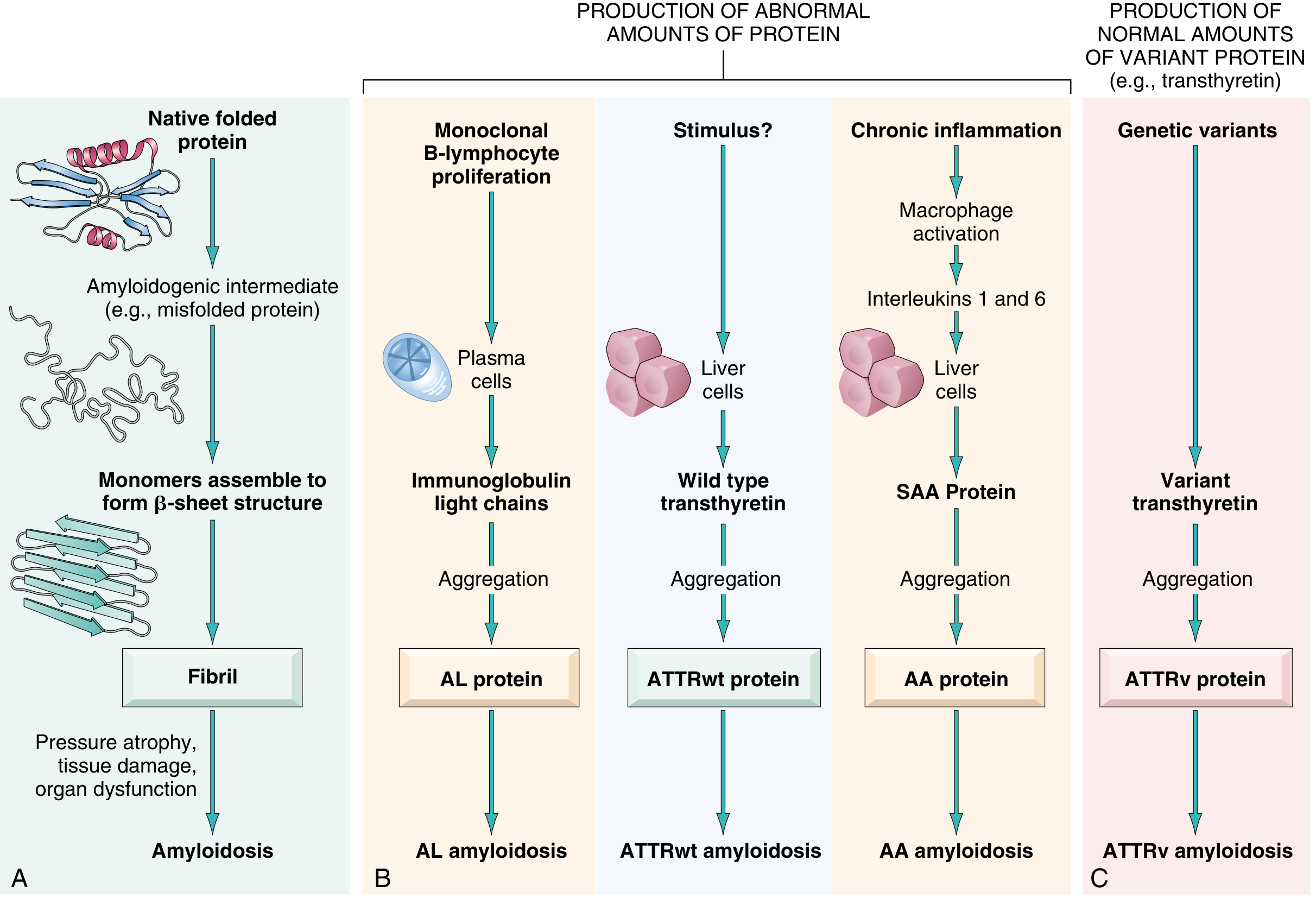
Fig. 6.45 - Robbins, Cotran & Kumar: Pathogenesis of amyloidosis showing the four main protein pathways
Classification Overview
Amyloidosis is broadly divided into Systemic (Generalized), Hereditary, and Localized forms.
I. Systemic (Generalized) Amyloidosis
1. AL Amyloidosis (Amyloid Light Chain)
- Previously called: Primary amyloidosis
- Fibril protein: Immunoglobulin light chains (chiefly λ type)
- Precursor: Ig light chains produced by clonal plasma cells
- Associated diseases: Multiple myeloma (5-15% develop amyloidosis), other monoclonal plasma cell proliferations, and monoclonal gammopathy
- Most common systemic form - ~2,000-3,000 new cases/year in the US
- Organs affected: heart, kidneys, liver, nerves, tongue, skin
- Key feature: Bence-Jones proteins (free κ or λ light chains) in serum and urine
2. ATTR Amyloidosis (Transthyretin)
Two subtypes:
- ATTRwt (wild-type): Formerly "senile systemic amyloidosis"; affects males >70 years; caused by age-related misfolding of normal transthyretin; predominantly cardiac
- ATTRv (variant/hereditary): Caused by mutant TTR gene; autosomal dominant; leads to familial amyloidotic polyneuropathy or cardiac amyloidosis depending on the TTR variant; a specific TTR variant is carried by ~4% of African-Americans and causes restrictive cardiomyopathy
3. AA Amyloidosis (Amyloid A)
- Previously called: Secondary/reactive amyloidosis
- Fibril protein: AA (serum amyloid A protein, SAA, produced by the liver)
- Mechanism: Chronic inflammation drives IL-6 and IL-1, stimulating sustained SAA production by the liver; only a subset of patients with elevated SAA develop amyloidosis
- Associated diseases: Rheumatoid arthritis, Crohn disease, ankylosing spondylitis, chronic infections (TB, osteomyelitis), hidradenitis suppurativa
- Organs mainly affected: kidneys, spleen, liver, adrenals
4. Aβ2M Amyloidosis (Dialysis-Related)
- Fibril protein: β-2 microglobulin
- Associated with: Long-term hemodialysis (β-2 microglobulin is normally filtered by kidneys and not adequately cleared by dialysis membranes)
- Deposits in joints, tendons, carpal tunnel
II. Hereditary (Familial) Amyloidosis
| Type | Fibril Protein | Notes |
|---|---|---|
| Familial Mediterranean Fever (FMF) | AA | Autosomal recessive; Armenian, Sephardic Jewish, Arabic origin; pyrin gene mutation; recurrent fevers, serositis |
| Familial Amyloidotic Polyneuropathy (FAP) | ATTRv | Autosomal dominant; mutant transthyretin; peripheral neuropathy predominant |
| Familial Cardiac Amyloidosis | ATTRv | Mutant TTR with cardiac predominance; common in African-Americans (Val122Ile variant) |
III. Localized Amyloidosis
Deposits confined to a single organ or tissue - no systemic spread.
| Location | Fibril Protein | Associated Condition |
|---|---|---|
| Brain (senile cerebral) | Aβ (amyloid beta) | Alzheimer disease; derived from APP (amyloid precursor protein) |
| Islets of Langerhans | AIAPP (amylin) | Type 2 diabetes mellitus |
| Thyroid C-cells | ACal (calcitonin-derived) | Medullary carcinoma of the thyroid |
| Isolated atrial | AANF (atrial natriuretic factor) | Isolated atrial amyloidosis |
| Skin, lung, larynx, bladder | AL (usually) | Nodular deposits, often with plasma cell infiltrates |
IV. Cutaneous (Primary Localized) Amyloidosis
A clinically distinct group from dermatology:
| Subtype | Features |
|---|---|
| Macular amyloidosis | Rippled hyperpigmented macules, mainly interscapular |
| Lichen amyloidosis | Pruritic lichenoid papules, shins |
| Nodular amyloidosis | Nodules, often on limbs/face; AL protein |
| Secondary (tumor-associated) | Amyloid deposits within skin tumors (e.g., basal cell carcinoma) |
| Familial primary cutaneous | Rare hereditary form |
| Pharmaceutical amyloidosis | Injection-site deposits |
Histological Identification (All Types)
- Congo red stain - positive; apple-green birefringence under polarized light (pathognomonic)
- PAS positive, diastase resistant
- Thioflavin T fluorescence
- Electron microscopy: straight, non-branching fibrils, 60-100 nm diameter
- Key distinction: AA amyloid loses Congo red birefringence after potassium permanganate treatment; AL and localized forms do not
Summary Table (Robbins Classification)
| Category | Fibril Protein | Precursor | Main Disease |
|---|---|---|---|
| AL amyloidosis | AL | Ig light chains (λ) | Myeloma, MGUS |
| ATTRwt amyloidosis | ATTRwt | Transthyretin (wild-type) | Aging males >70 |
| AA amyloidosis | AA | SAA | Chronic inflammation |
| ATTRv amyloidosis | ATTRv | Transthyretin (variant) | Familial neuropathy/cardiomyopathy |
| FMF | AA | SAA | Familial Mediterranean fever |
| Alzheimer | Aβ | APP | Senile cerebral |
| Type 2 DM | AIAPP | Islet amyloid peptide | Pancreatic islets |
| Medullary thyroid Ca | ACal | Calcitonin | Thyroid |
| Atrial | AANF | Atrial natriuretic factor | Isolated atrial |
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Table 6.17, Fig. 6.45)
- Robbins & Kumar Basic Pathology
- Andrews' Diseases of the Skin (p. 56-94)
- Dermatology 2-Volume Set 5e
This is a shared conversation. Sign in to Orris to start your own chat.