Hemostasis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Excellent content gathered. I now have everything needed for a comprehensive response. Let me compile it.
Hemostasis
Hemostasis is a precisely orchestrated process involving platelets, coagulation factors, and the endothelium that occurs at a site of vascular injury to form a blood clot and prevent excessive bleeding. It can be divided into four sequential but overlapping phases.
1. Vascular Spasm
Immediately after injury, arteriolar vasoconstriction reduces blood flow to the injured area. This is mediated by neurogenic reflexes and amplified by local release of endothelin, a potent vasoconstrictor from damaged endothelium. The effect is transient — bleeding resumes without platelet and coagulation activation. — Robbins & Kumar Basic Pathology
2. Primary Hemostasis — The Platelet Plug
Platelets are anucleate disc-shaped fragments shed from bone marrow megakaryocytes (differentiation regulated by thrombopoietin); they circulate for ~7–10 days with 20–30% stored in the spleen.
Platelet Adhesion
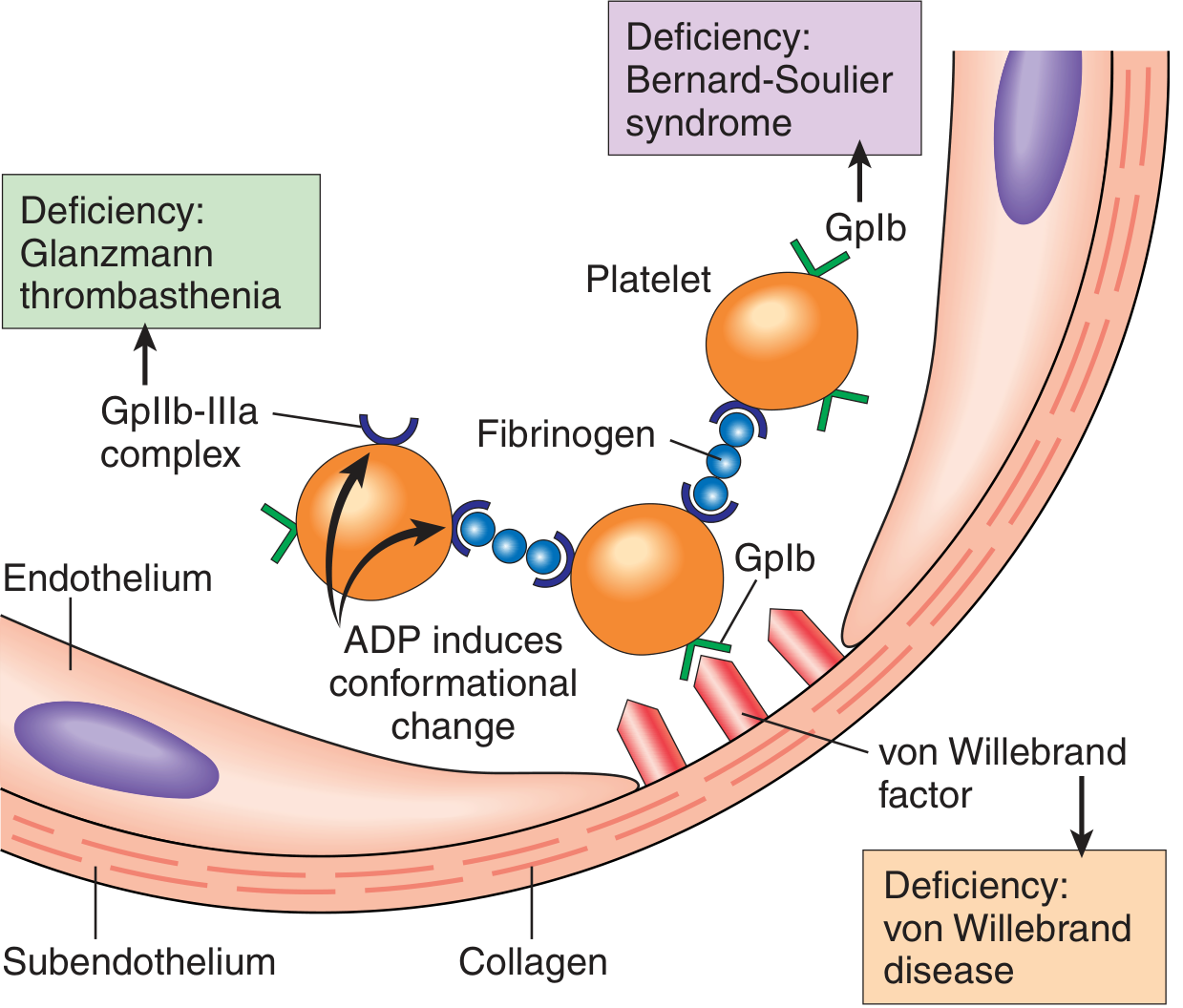
Disruption of endothelium exposes subendothelial collagen, which binds von Willebrand factor (vWF). At high shear stress, vWF bridges between collagen and GpIb (part of the platelet GPIb/V/IX complex). Stable adhesion involves direct collagen binding via GPVI and GPIa/IIa (integrin α2β1). Deficiency of vWF → von Willebrand disease; deficiency of GpIb → Bernard-Soulier syndrome. — Robbins & Kumar; Tietz Textbook of Laboratory Medicine
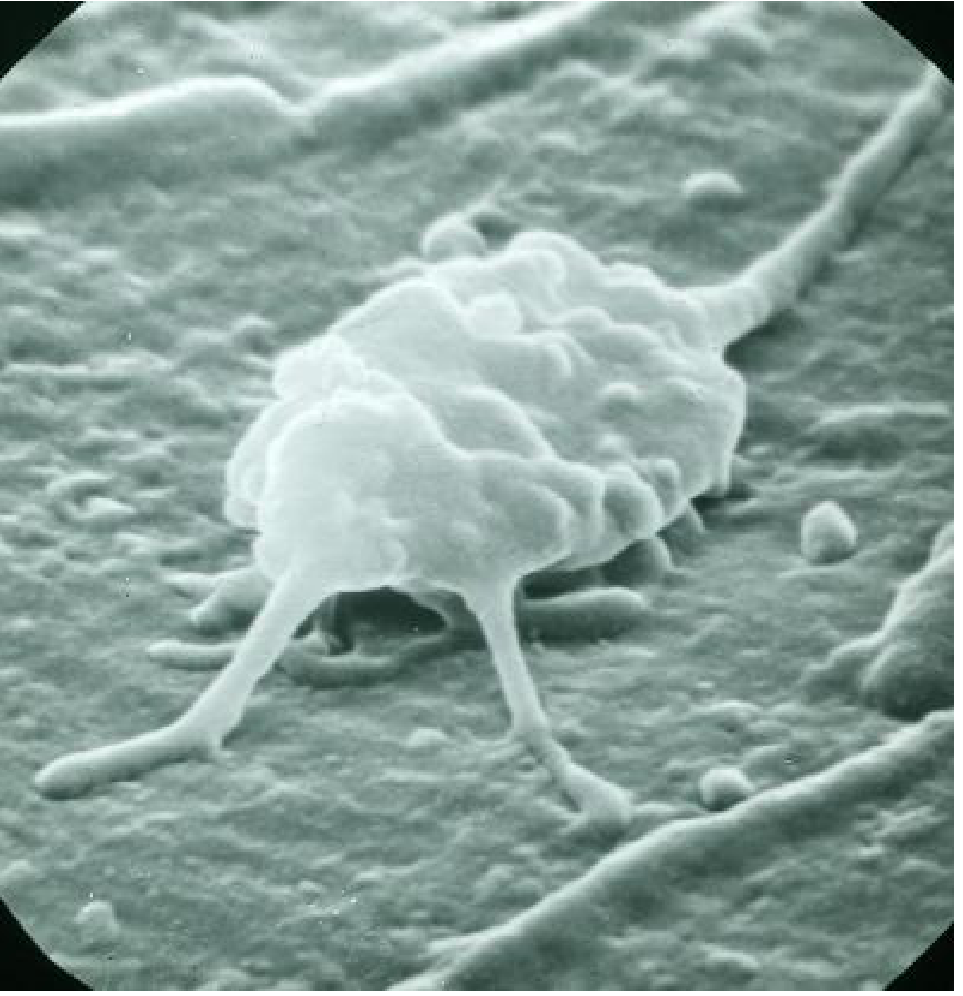
Platelet Activation (Shape Change + Granule Release)
Adherent platelets convert from smooth discs to spiky "sea urchins" with filopodia (greatly increasing surface area). This shape change:
- Translocates phosphatidylserine to the outer leaflet — a negatively charged surface that nucleates coagulation factor complexes
- Increases GpIIb/IIIa affinity for fibrinogen
Granule secretion (release reaction):
| Granule Type | Contents |
|---|---|
| α-granules | Fibrinogen, factor V, vWF, fibronectin, platelet factor 4, PDGF, TGF-β, P-selectin |
| Dense (δ) granules | ADP, ATP, Ca²⁺, serotonin, epinephrine, polyphosphate |
Key activators include thrombin (via protease-activated receptors, PARs) and ADP (via P2Y1/P2Y12 receptors). Activated platelets synthesize thromboxane A₂ (TxA₂) — a potent aggregator and vasoconstrictor. Aspirin irreversibly inhibits cyclooxygenase, blocking TxA₂ synthesis.
Platelet Aggregation
ADP and TxA₂ recruit additional platelets. Fibrinogen cross-links adjacent platelets via GpIIb/IIIa (integrin αIIbβ3), forming the primary hemostatic plug. Deficiency of GpIIb/IIIa → Glanzmann thrombasthenia.
3. Secondary Hemostasis — The Coagulation Cascade
Secondary hemostasis consolidates the platelet plug by depositing insoluble fibrin around it.
General Principles
Each reaction in the cascade involves:
- An enzyme (activated coagulation factor, usually a serine protease)
- A substrate (inactive proenzyme)
- A cofactor (reaction accelerator)
These complexes assemble on the negatively charged phospholipid surface of activated platelets, with calcium holding components together. Calcium binding requires γ-carboxylation of glutamic acid residues on factors II, VII, IX, X — a reaction dependent on vitamin K (antagonized by warfarin).
The Cascade — Lab vs. In Vivo
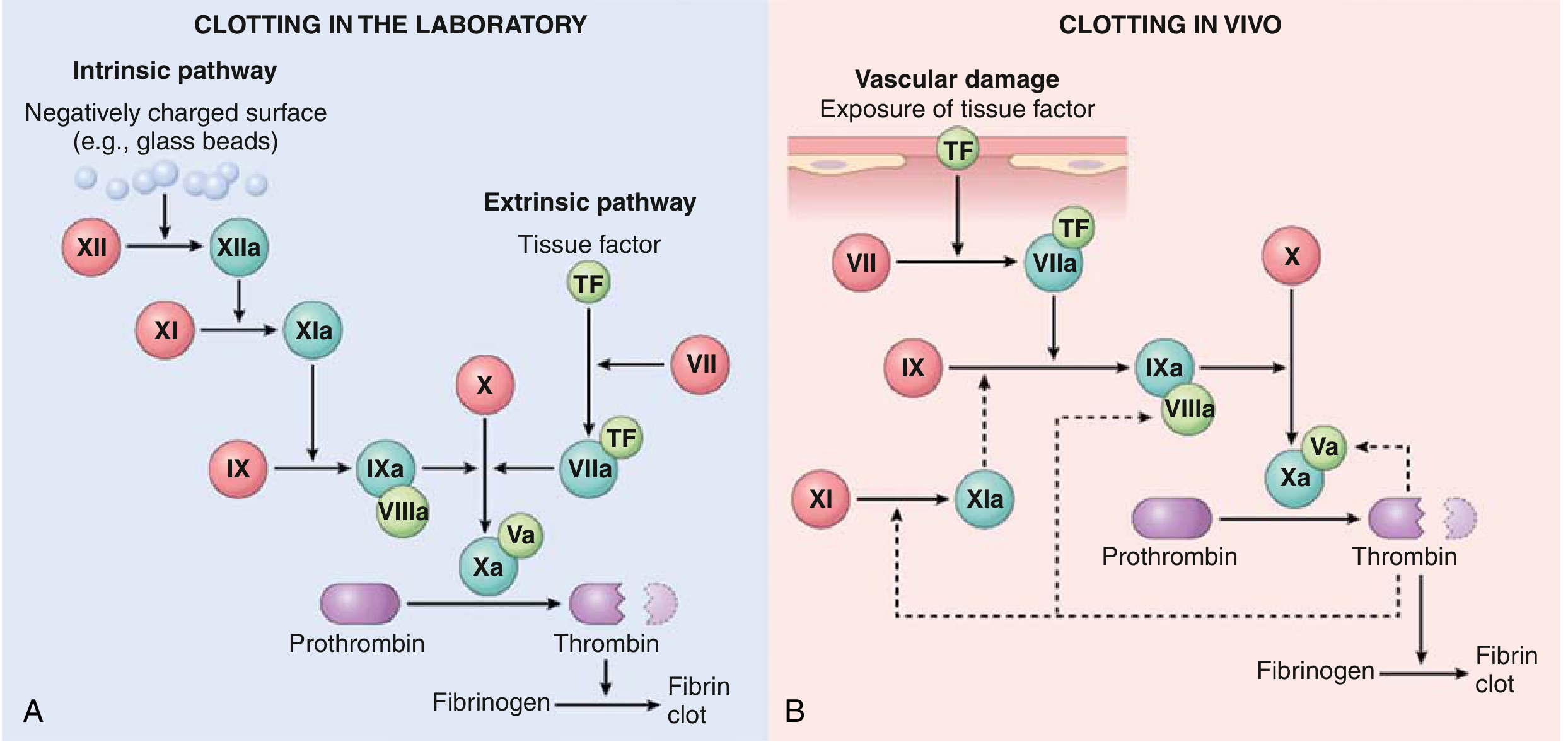
| Pathway | Lab Test | Factors Assessed |
|---|---|---|
| Extrinsic | PT (Prothrombin Time) | TF, VII, X, V, II, fibrinogen |
| Intrinsic | PTT (Partial Thromboplastin Time) | XII, XI, IX, VIII, X, V, II, fibrinogen |
| Common | Both | X, V, II, fibrinogen |
In vivo, the TF/VIIa complex is the dominant initiator — it activates factor IX, and the IXa/VIIIa complex (intrinsic tenase) is the major activator of factor X. Factor XII deficiency causes no bleeding (physiologic role uncertain), while factor XI deficiency causes only mild bleeding (thrombin can activate XI as a feedback amplifier).
Thrombin — The Central Mediator
| Thrombin Action | Effect |
|---|---|
| Cleaves fibrinogen → fibrin monomers | Forms insoluble fibrin mesh |
| Activates factors V, VIII, XI | Amplifies the cascade |
| Activates factor XIII | Covalently cross-links fibrin |
| Activates PARs on platelets | Potent platelet activator |
| Activates PARs on endothelium | Promotes inflammation/repair |
| Binds thrombomodulin → activates Protein C | Anticoagulant switch |
Vitamin K-dependent factors: II (prothrombin), VII, IX, X, and anticoagulants Protein C and Protein S.
4. Clot Stabilization
- Factor XIII (a transglutaminase activated by thrombin) covalently cross-links fibrin polymers
- Platelet contractile filopodia retract, drawing wound edges together (clot retraction)
- The result is a solid permanent plug
Regulation of Coagulation — Limiting Clot Propagation
Without counterregulatory mechanisms, coagulation would spread systemically. The key brakes:
Anticoagulant Mechanisms of Endothelium
| Mechanism | Mediator | Target |
|---|---|---|
| PGI₂ (prostacyclin) | Endothelium | Inhibits platelet aggregation; vasodilator |
| Nitric oxide (NO) | Endothelium | Inhibits platelet activation; vasodilator |
| Adenosine diphosphatase | Endothelium | Degrades ADP |
| Antithrombin (AT-III) | Plasma (heparin-like compounds on endothelium) | Inhibits thrombin, Xa, IXa, XIa |
| Thrombomodulin + Protein C/S | Endothelium | Inactivates Va and VIIIa |
| TFPI | Endothelium | Inhibits TF/VIIa and Xa (stops initiation) |
Protein C pathway: Thrombin binds thrombomodulin on endothelium → activates Protein C (with cofactor Protein S) → cleaves/inactivates factors Va and VIIIa. Deficiency of Protein C or S → hypercoagulable state. — Tietz; Robbins
5. Fibrinolysis — Clot Dissolution
Fibrinolysis dissolves the clot to allow tissue healing:
- Plasminogen (inactive circulating precursor) binds fibrin within the clot
- t-PA (tissue plasminogen activator), synthesized by endothelium, is maximally active when bound to fibrin — confining lytic activity to the clot
- t-PA cleaves plasminogen → plasmin, which degrades fibrin
- D-dimers (fibrin degradation products) are released — clinically useful markers of thrombosis (e.g., DVT, PE, DIC)
Regulation of fibrinolysis:
- α₂-antiplasmin — rapidly inhibits free circulating plasmin
- PAI-1 (plasminogen activator inhibitor-1) — endothelial inhibitor of t-PA
- TAFI (thrombin-activatable fibrinolysis inhibitor) — activated by thrombin/thrombomodulin complex, inhibits fibrinolysis
Summary Diagram of Hemostatic Sequence
| Phase | Key Components | Outcome |
|---|---|---|
| Vascular spasm | Endothelin, neurogenic reflex | ↓ Blood flow |
| Primary hemostasis | Platelets, vWF, GpIb, GpIIb/IIIa, ADP, TxA₂ | Platelet plug |
| Secondary hemostasis | TF, coagulation cascade, thrombin | Fibrin mesh |
| Clot stabilization | Factor XIII, platelet contraction | Permanent plug |
| Fibrinolysis | Plasmin, t-PA | Clot dissolution |
Clinical Correlates
| Disorder | Defect |
|---|---|
| von Willebrand disease | ↓ vWF → impaired platelet adhesion |
| Bernard-Soulier syndrome | GpIb deficiency → no vWF binding |
| Glanzmann thrombasthenia | GpIIb/IIIa deficiency → no fibrinogen bridging |
| Hemophilia A | Factor VIII deficiency → ↑ PTT |
| Hemophilia B | Factor IX deficiency → ↑ PTT |
| Warfarin | Blocks vitamin K → ↓ II, VII, IX, X → ↑ PT/INR |
| Heparin | Potentiates antithrombin → inhibits IIa/Xa |
| DIC | Systemic activation → consumption of factors + fibrinolysis |
| Cirrhosis | ↓ Pro- and anticoagulant factors → "rebalanced" hemostasis |
Sources: Robbins & Kumar Basic Pathology; Tietz Textbook of Laboratory Medicine, 7th ed.; Miller's Anesthesia, 10th ed.
This is a shared conversation. Sign in to Orris to start your own chat.