Tell me about renal ccell carcinoma renal oncytoma nephroblastoma urothelisl carcinoma of bladder complete concept with every single detail in paragraph
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the material needed for a comprehensive response. Let me write the full detailed answer.
Renal Cell Carcinoma, Renal Oncocytoma, Nephroblastoma, and Urothelial Carcinoma of the Bladder
1. RENAL CELL CARCINOMA (RCC)
Epidemiology and Overview
Renal cell carcinomas arise from the renal tubular epithelium and are located predominantly in the cortex. They account for 80% to 85% of all primary malignant neoplasms of the kidney and represent 2% to 3% of all cancers in adults, with approximately 65,000 new cases per year in the United States; nearly 40% of patients die of the disease. These tumors are most common in the sixth and seventh decades of life, and men are affected about twice as often as women. Risk factors include cigarette smoking (which doubles incidence), obesity (particularly in females), hypertension, unopposed estrogen therapy, and occupational exposure to cadmium, asbestos, and petroleum products. The risk is dramatically increased - up to 30-fold - in individuals with acquired polycystic disease complicating chronic dialysis, and there is also an increased risk in patients with end-stage kidney disease, tuberous sclerosis, and chronic kidney disease. About 15% of patients have distant metastases at first presentation.
Familial Syndromes
Although most renal cancers are sporadic, several autosomal dominant familial syndromes exist and have provided enormous insight into renal carcinogenesis. In von Hippel-Lindau (VHL) syndrome, one-half to two-thirds of affected individuals develop bilateral, often multiple, clear cell carcinomas. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome is caused by loss-of-function mutations in the FH gene (fumarate hydratase), resulting in cutaneous and uterine leiomyomata alongside an aggressive high-grade papillary carcinoma with early metastatic spread; these are now called fumarate hydratase-deficient RCCs. Hereditary papillary carcinoma is an autosomal dominant form with multiple bilateral papillary tumors and low nuclear grade, driven by germline gain-of-function MET mutations. Birt-Hogg-Dube (BHD) syndrome results from mutations in the BHD gene encoding the tumor suppressor folliculin, presenting with fibrofolliculomas of the skin, pulmonary cysts, and renal tumors of multiple subtypes.
Classification and Pathogenesis
RCC is classified into three main subtypes based on cytogenetics, genetics, and histology.
Clear Cell Carcinoma is the most common type, accounting for 65-80% of renal cell carcinomas. The molecular hallmark is loss or inactivation of both copies of the VHL gene on chromosome 3p25. In VHL disease (an autosomal dominant disorder predisposing to hemangioblastomas of the cerebellum and retina), bilateral clear cell RCCs develop in 40-60% of individuals. Sporadic cases also involve monoallelic deletion of chromosome 3p carrying the VHL gene plus mutation or silencing of the second allele by somatic mutation or hypermethylation. In 98% of clear cell RCCs, there is a deletion at chromosome 3p25.3. The VHL protein normally targets hypoxia-inducible factor (HIF-1) for oxygen-dependent degradation via a ubiquitin ligase complex. When VHL is inactive, HIF-1 levels remain high even under normoxic conditions, causing inappropriate expression of VEGF (promoting tumor angiogenesis), insulin-like growth factor-1 (IGF-1, stimulating growth), and other HIF target genes. HIF also collaborates with MYC to reprogram cellular metabolism toward growth. Additionally, deep sequencing has revealed frequent loss-of-function mutations in genes encoding chromatin-remodeling proteins that regulate histone methylation, demonstrating that epigenomic dysregulation plays a central role.
Papillary Renal Cell Carcinoma accounts for 10-15% of renal cancers and occurs in both familial and sporadic forms. Unlike clear cell RCC, it is not associated with chromosome 3p deletions. The unifying molecular abnormality is gain of function of the MET proto-oncogene, a tyrosine kinase receptor on chromosome 7q for hepatocyte growth factor (scatter factor). In the familial form, germline activating MET mutations drive abnormal growth of proximal tubular epithelial cells. Sporadic tumors show increased MET copy number from trisomies of chromosome 7 (and 17) or somatic MET mutations. Papillary RCC tends to be multifocal and bilateral, frequently presenting as early-stage tumors.
Chromophobe Renal Cell Carcinoma is the least common of the three main types, representing about 5% of RCCs. It arises from the intercalated cells of the collecting ducts. The tumor cells are pale (chromophobe) with lightly eosinophilic cytoplasm but do not appear clear as in clear cell carcinoma. These neoplasms frequently display multiple losses of entire chromosomes leading to extreme hypoploidy. The critical oncogenic drivers have not been fully determined, but in general chromophobe RCC carries a favorable prognosis compared to the other subtypes.
Gross Morphology
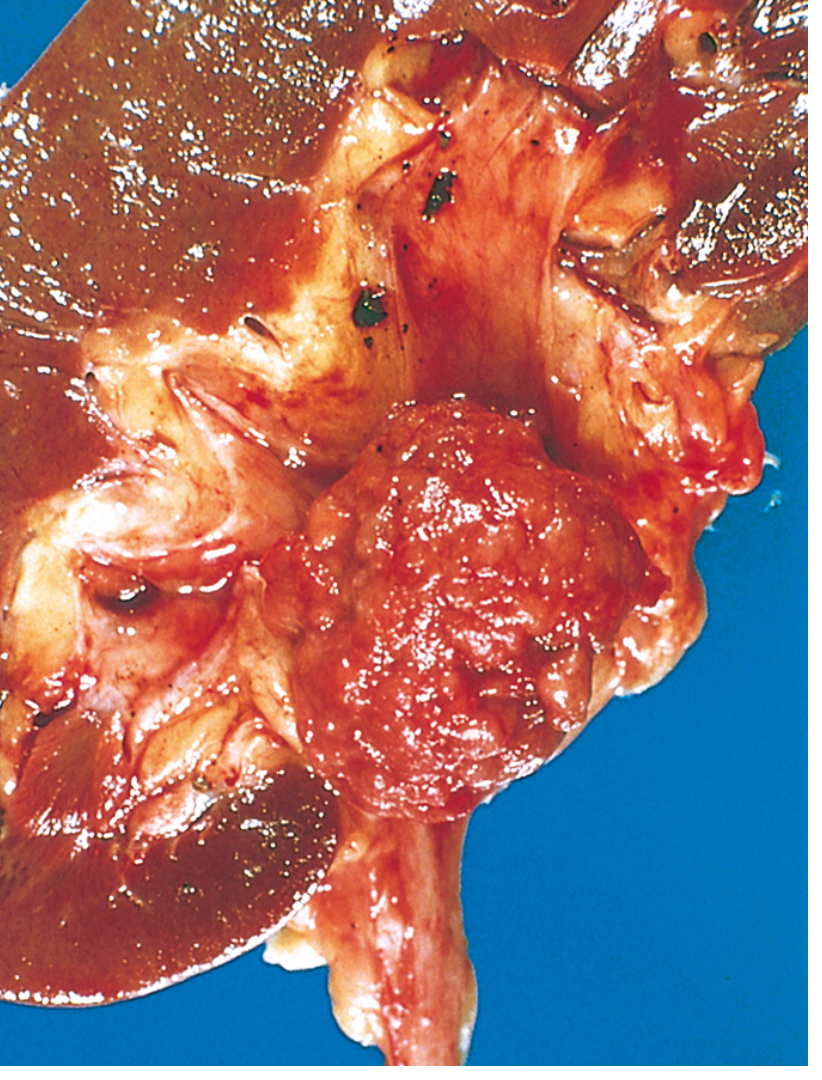
Clear cell carcinomas are typically large, solitary, spherical masses ranging from 3 to 15 cm in diameter when symptomatic. The cut surface is characteristically yellow to orange due to abundant intracellular lipid and glycogen, with areas of necrosis, cystic change, and hemorrhage. The margins are usually well defined. As the tumor enlarges, it may fungate through the walls of the collecting system into the calyces and pelvis as far as the ureter. One of the most clinically important features is the tumor's tendency to invade the renal vein and grow as a solid column within the vessel, sometimes extending in serpentine fashion into the inferior vena cava and even the right side of the heart. Direct invasion into the perinephric fat and adrenal gland may also be seen.
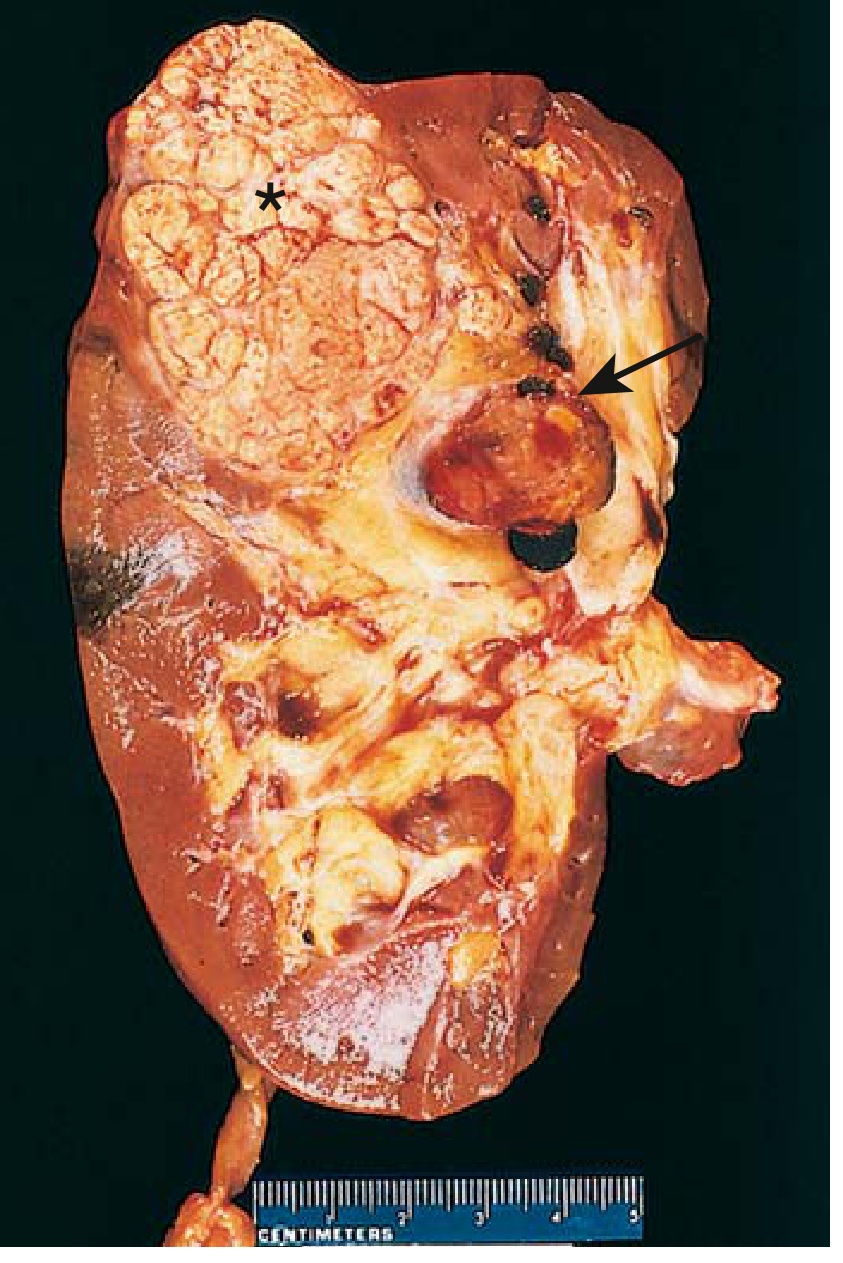
Renal cell carcinoma. Typical cross-section showing yellowish neoplasm (asterisk) with tumor in the dilated thrombosed renal vein (arrow). - Robbins & Kumar Basic Pathology
Histology
In clear cell type (Fig. A below), most neoplastic cells are arranged in nests or alveoli separated by a delicate fibrovascular stroma. Cells have clear vacuolated cytoplasm with distinct cell membranes due to abundant lipid and glycogen, and small round nuclei. In papillary type (Fig. B), the tumor shows characteristic papillary or tubulopapillary formations, often with foamy macrophages in the fibrovascular stalks. In chromophobe type (Fig. C), the cells are large with pale to eosinophilic granular cytoplasm arranged in solid sheets, and the cell membranes are prominent.
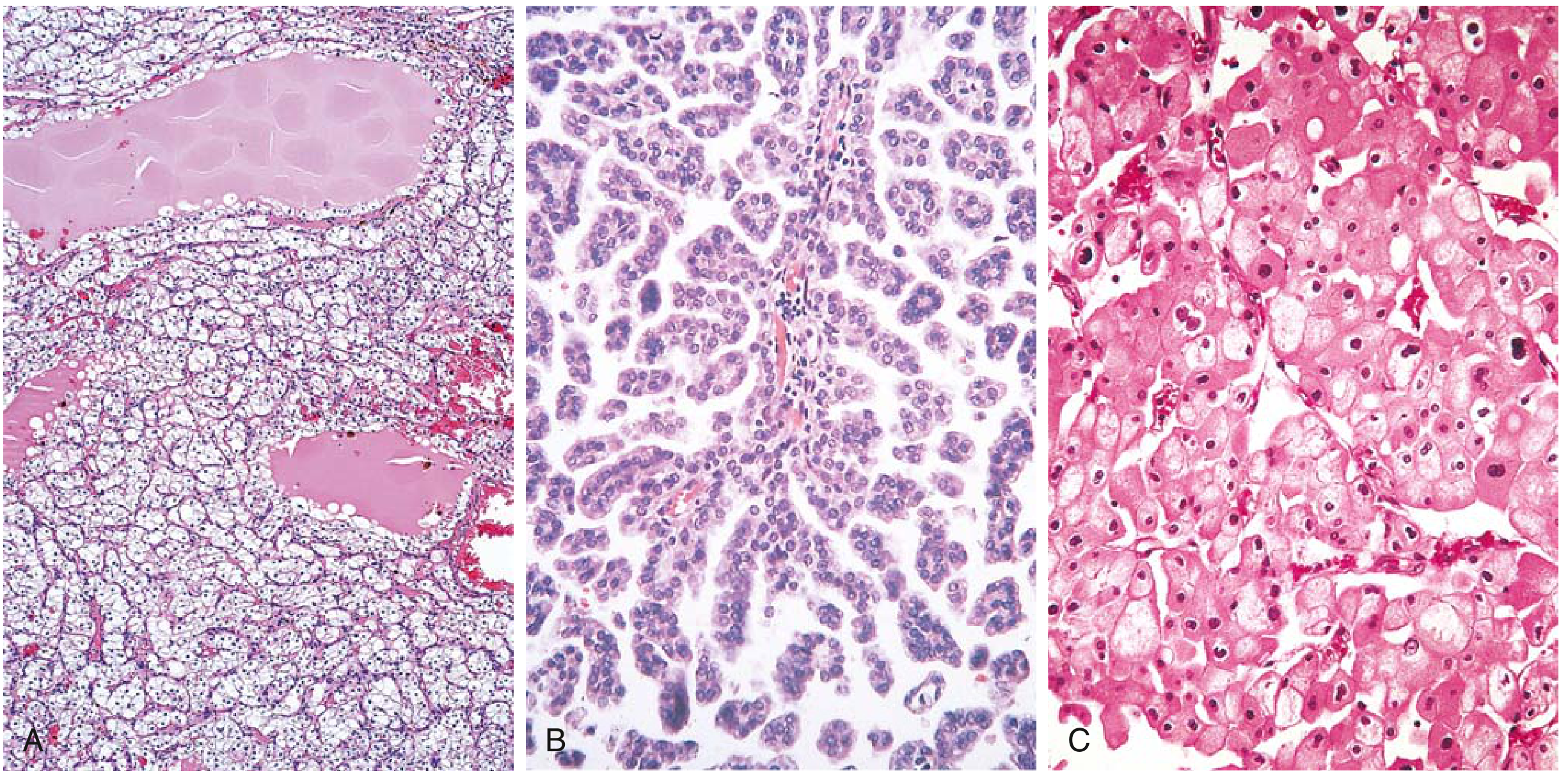
RCC histological subtypes. (A) Clear cell type. (B) Papillary type with foamy macrophages in stalks. (C) Chromophobe type. - Robbins & Kumar Basic Pathology
Clinical Features
RCC is notorious for its wide variety of clinical presentations and is rightly called "the great mimic in medicine." The classic triad of painless hematuria, flank pain, and a palpable abdominal mass is found in only 10% of patients at presentation. Most cases today are discovered incidentally on imaging performed for other reasons. The tumor produces diverse systemic syndromes through abnormal hormone secretion, including polycythemia (from erythropoietin), hypercalcemia (ectopic PTHrP), hypertension, hepatic dysfunction (Stauffer syndrome), feminization or masculinization, Cushing syndrome (ectopic ACTH), eosinophilia, leukemoid reactions, and amyloidosis. A particularly troublesome feature is the tumor's propensity to metastasize widely before producing any local symptoms. The most common sites of metastasis are the lungs (>50%), bones (33%), followed by regional lymph nodes, liver, adrenal glands, and brain.
Imaging
On CT and MRI, clear cell RCC demonstrates avid enhancement in the corticomedullary phase and becomes hypoenhancing in the nephrographic phase due to its rich vascularity from VEGF-driven angiogenesis. Chromophobe RCC also shows avid enhancement in the corticomedullary phase but to a lesser degree. Papillary RCC is characteristically hypointense on T2-weighted MRI and hypoenhancing on all postcontrast phases. MRI is more sensitive than CT for identifying complex cystic features (wall thickening, septations) and detecting enhancement through subtraction imaging. The Bosniak classification system stratifies renal cysts by CT/MRI findings into categories I-IV, guiding surgical versus surveillance decisions.
Treatment and Prognosis
The primary treatment for localized RCC is surgery. Nephron-sparing surgery (partial nephrectomy) is recommended for T1a tumors (<4 cm) and increasingly for larger tumors when technically feasible, in order to preserve renal function. Radical nephrectomy is performed for larger or more complex tumors. Even when the tumor invades the renal vein or inferior vena cava, surgical excision can be curative if no distant metastases are present. For metastatic disease, systemic therapy has dramatically evolved: drugs targeting the VEGF pathway (sunitinib, pazopanib, axitinib, bevacizumab) and mTOR inhibitors (everolimus, temsirolimus) are used, as are immune checkpoint inhibitors (nivolumab, pembrolizumab), often in combination. The average 5-year survival is about 70% overall and approaches 100% in the absence of distant metastases. With renal vein invasion or perinephric fat extension, 5-year survival drops to approximately 60%. With distant metastases, prognosis is significantly worse but immunotherapy combinations have improved outcomes considerably. - Robbins & Kumar Basic Pathology, p. 531-532; Robbins Cotran & Kumar Pathologic Basis of Disease, p. 879-882
2. RENAL ONCOCYTOMA
Definition and Cell of Origin
Renal oncocytoma is a benign epithelial neoplasm of the kidney. It is thought to arise from the intercalated cells of the collecting ducts and accounts for approximately 3-7% of all renal neoplasms. It is the most common benign solid renal tumor in adults.
Pathogenesis and Molecular Features
The hallmark of oncocytoma is an abundance of mitochondria within tumor cells - this is the direct basis for their characteristic finely granular eosinophilic cytoplasm visible on light microscopy, and their tan or mahogany brown color on gross examination. Ultrastructurally, the cytoplasm of oncocytoma cells is packed with mitochondria (as seen in the electron micrograph image below). The tumor cells harbor disruptive mutations that lead to loss of Complex I, a key component of the mitochondrial electron transport chain required for oxidative phosphorylation. This impairment activates feedback loops that paradoxically increase mitochondrial proliferation, accounting for the characteristic morphology. Chromosomal abnormalities are present, including loss of chromosomes 1 and Y, and rearrangements involving the cyclin D1 locus. Multiple oncocytomas may be seen in patients with tuberous sclerosis (referred to as renal oncocytosis), and in familial cases where tumors are multifocal.
Gross Morphology
Grossly, oncocytomas appear as tan or mahogany brown, relatively homogeneous, well-circumscribed masses. A central stellate scar is present in about one-third of cases and is considered a helpful (though not pathognomonic) feature distinguishing them from RCC on imaging. Despite their benign nature, they may reach large sizes - up to 12 cm in diameter.
Histology
On light microscopy, the tumor is composed of uniform cells with abundant granular eosinophilic cytoplasm and small, round, benign-appearing nuclei with small but conspicuous nucleoli. The cells are arranged in nests or tubules within a loose stroma. There is no nuclear atypia, necrosis, or mitotic activity typical of malignancy.
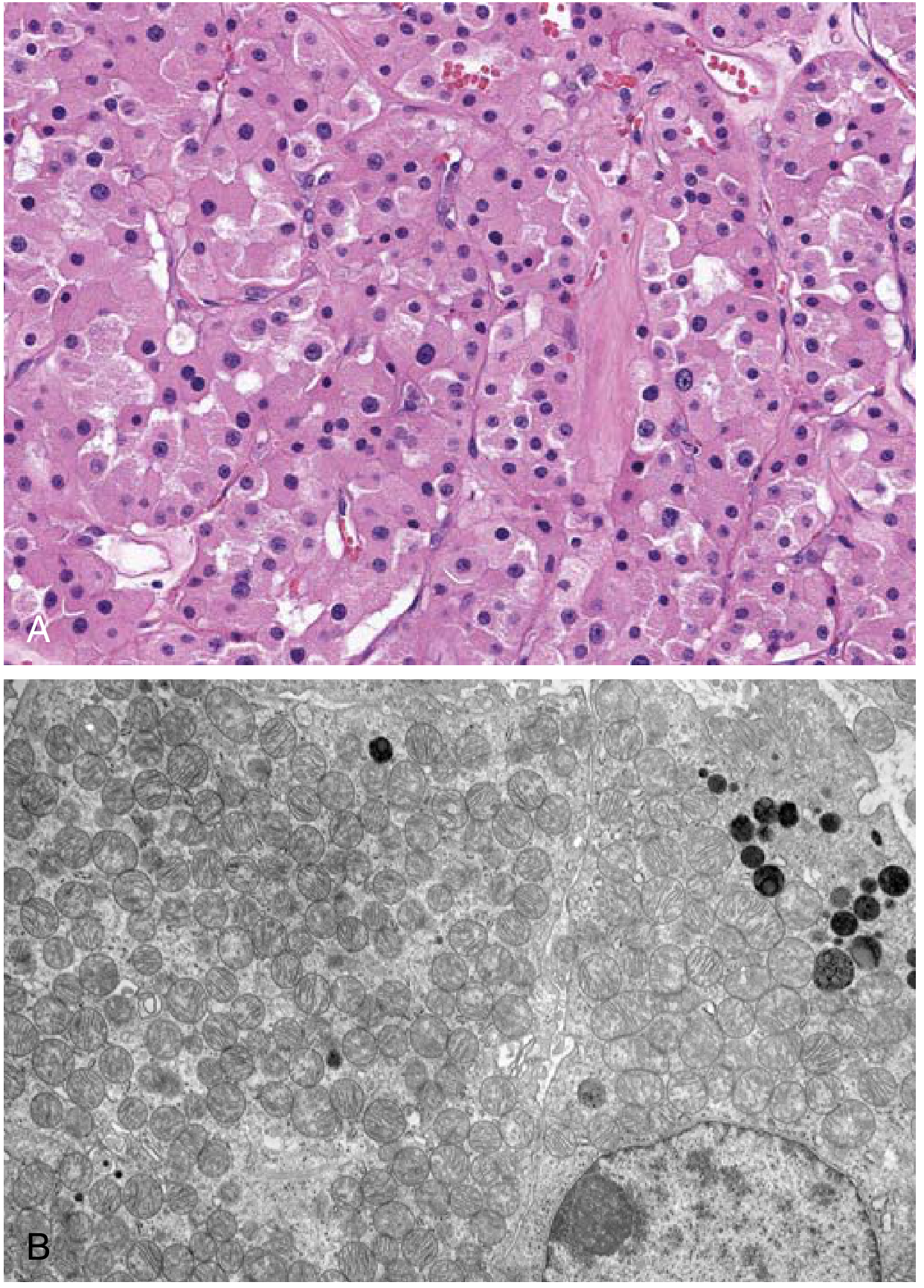
Renal oncocytoma. (A) Uniform cells with granular eosinophilic cytoplasm. (B) Electron micrograph showing the cytoplasm packed with mitochondria. - Robbins & Kumar Basic Pathology
Diagnostic Challenge
The most clinically important aspect of oncocytoma is that it is often difficult - and sometimes impossible - to distinguish it from chromophobe RCC on imaging alone, and even on fine-needle biopsy. Chromophobe RCC also has eosinophilic granular cells and can share many imaging characteristics. The central scar and typical tan color help, but are not definitive. In 10-30% of patients with multiple oncocytic nodules (oncocytosis), there may be a coexisting renal cell carcinoma, requiring careful monitoring. Because preoperative distinction can be unreliable, many oncocytomas are resected surgically and only then confirmed as benign. Partial nephrectomy is the preferred approach for suspected oncocytoma. - Robbins & Kumar Basic Pathology, p. 533; Robbins Cotran & Kumar Pathologic Basis of Disease, p. 878-879; Smith and Tanagho's General Urology, 19e
3. NEPHROBLASTOMA (WILMS TUMOR)
Epidemiology and Overview
Nephroblastoma, universally known as Wilms tumor, is the most common solid renal tumor of childhood and ranks as the third most common solid non-hematologic malignancy in children under 10 years of age. It accounts for approximately 5% of all childhood cancers, with roughly 650 new cases reported annually in the United States. The peak age at presentation is the third year of life (around 3.5 years), and there is no sex predilection. The disease is seen worldwide with a similar age of onset and sex distribution. Tumors are most commonly unicentric and can arise in either kidney with equal frequency; 5% of cases are bilateral. Approximately 10% of patients have associated congenital malformations.
Associated Syndromes
Wilms tumor occurs in both sporadic and familial forms. The familial form accounts for approximately 1% of cases and is inherited as an autosomal dominant trait. Several important congenital syndromes are associated with increased Wilms tumor risk:
- WAGR syndrome (Wilms tumor, Aniridia, Genitourinary malformations, and intellectual disability/mental Retardation): caused by deletions at chromosome 11p13 that encompass the WT1 gene.
- Beckwith-Wiedemann syndrome: an overgrowth syndrome characterized by macroglossia, visceromegaly, and hemihypertrophy, associated with alterations in the IGF1, H19, and p57 genes (linked to chromosome 11p15 imprinting).
- Isolated hemihypertrophy: associated with increased risk even without other features of Beckwith-Wiedemann.
- Denys-Drash syndrome: WT1 mutation causing gonadal dysgenesis, nephropathy, and Wilms tumor.
- Genitourinary abnormalities (hypospadias, cryptorchidism, renal fusion) are found in 4.5-7.5% of patients with unilateral Wilms tumor and up to 13.4% of those with bilateral disease.
Pathogenesis and Genetics
Knudson and Strong's two-hit hypothesis applies directly to Wilms tumor. In familial cases, one germline mutation is inherited, and a single subsequent somatic ("second hit") mutation in the affected kidney is sufficient to initiate tumorigenesis, explaining the earlier onset and bilateral/multifocal presentation. In sporadic cases, both mutations must occur post-zygotically in the same cell, making tumorigenesis less probable but not rare. The WT1 gene (Wilms Tumor gene 1) was mapped to chromosome 11p13 and encodes a zinc finger transcription factor critical for normal kidney and gonadal development. Despite its discovery, only 5-10% of sporadic Wilms tumors have WT1 mutations, indicating that other genes (WT2 at 11p15, p53, and others) are also involved.
Precursor Lesions: Nephrogenic Rests
Beckwith and colleagues identified nephrogenic rests (NRs) as Wilms tumor precursor lesions - these are abnormally persistent embryonic renal tissue present after the 36th week of gestation. Two types are defined: perilobar NRs (found at the periphery of the renal lobe) and intralobar NRs (found within the parenchyma). These rests may remain dormant for years, undergo involution, or progress to form Wilms tumors. Nephroblastomatosis refers to diffuse or multifocal NRs and is associated with an increased risk of Wilms tumor development.
Pathology and Histology
The typical Wilms tumor is a triphasic neoplasm composed of three elements in varying proportions: blastema (primitive small round blue cells with hyperchromatic nuclei), epithelium (tubular and glomeruloid structures), and stroma (spindle cells, often with skeletal muscle differentiation). Tumors composed of blastema and stroma alone, or pure tubular/papillary forms, are also described.
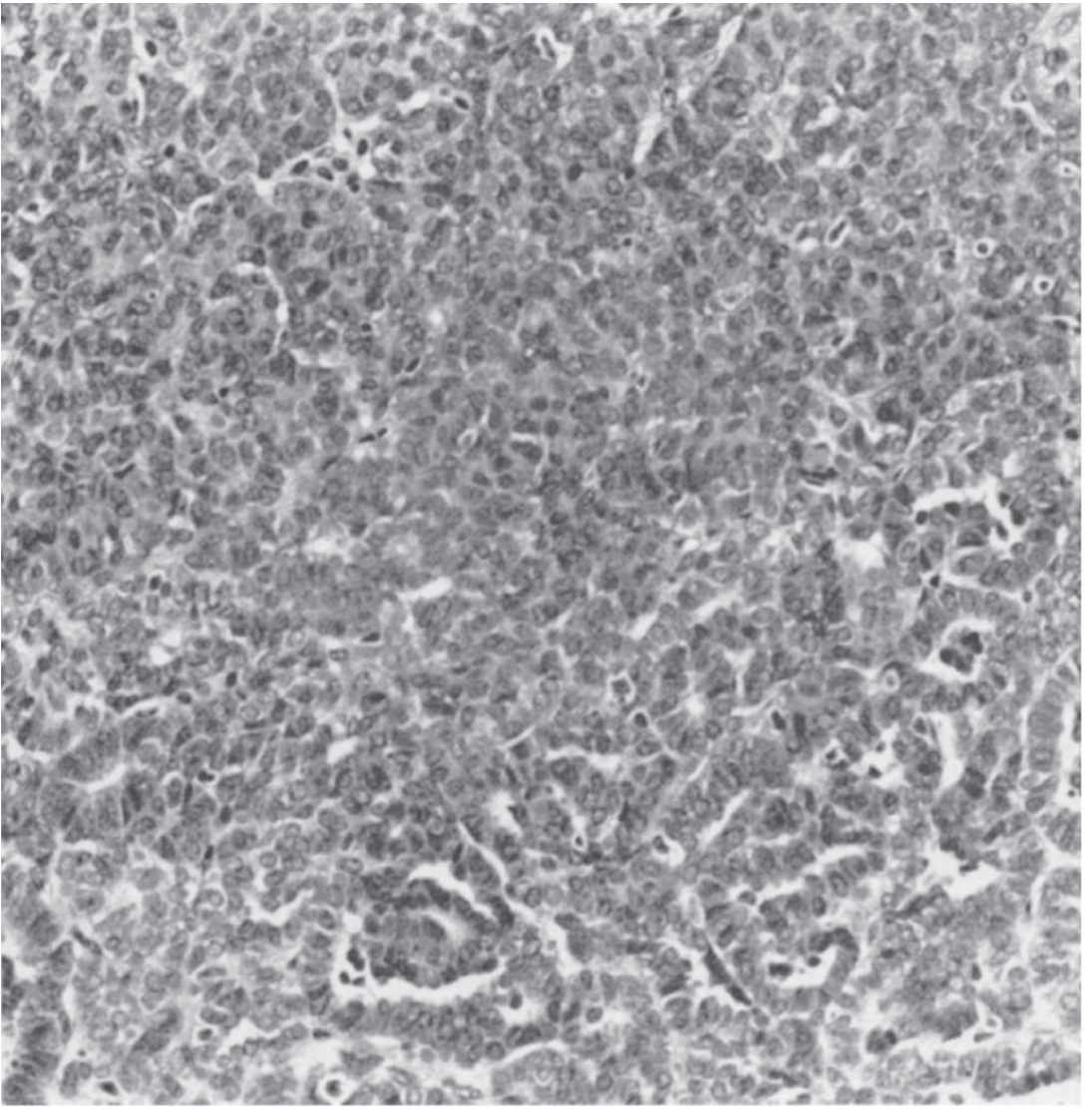
Wilms tumor with characteristic tubular/glomeruloid structures and blastema (original magnification x40). - Smith and Tanagho's General Urology, 19e
Grossly, Wilms tumors are typically large, multilobulated, and gray or tan in color with focal areas of hemorrhage and necrosis. A fibrous pseudocapsule is occasionally present. Tumor dissemination occurs by direct extension through the renal capsule, hematogenously via the renal vein and inferior vena cava, or via lymphatics. Metastatic disease at diagnosis occurs in 10-15% of patients, with lungs (85-95%) and liver (10-15%) being the most common metastatic sites. Regional lymph nodes are involved in up to 25% of patients.
Histologic Prognosis: Favorable vs. Unfavorable
The National Wilms Tumor Study (NWTS) Group divided histologic features into prognostically important groups. Favorable histology encompasses all Wilms tumors without anaplasia. Unfavorable histology includes tumors with focal or diffuse anaplasia (characterized by extreme nuclear atypia, hyperdiploidy, and complex chromosomal translocations), as well as two non-Wilms malignancies - clear cell sarcoma of the kidney and rhabdoid tumor of the kidney. Anaplasia occurs in approximately 5% of Wilms tumors; its incidence increases with age, is more common in African-American children, and is linked to p53 mutations. Diffuse anaplasia confers a significantly worse prognosis than focal anaplasia.
Staging (NWTS System)
- Stage I: Tumor limited to the kidney and completely excised; no capsule penetration or renal sinus vessel involvement.
- Stage II: Tumor extends beyond the kidney but is completely removed; may involve capsule penetration, renal sinus vessel invasion, local biopsy, or local spillage.
- Stage III: Residual non-hematogenous tumor confined to the abdomen (e.g., positive lymph nodes, peritoneal contamination, incomplete resection, or tumor spillage not confined to the flank).
- Stage IV: Hematogenous metastases (lung, liver, bone, brain) or lymph node metastases beyond the abdomino-pelvic region.
- Stage V: Bilateral renal involvement at diagnosis.
Clinical Presentation
Children most commonly present with a smooth, firm, non-tender abdominal mass discovered by a parent or during routine examination. Abdominal pain, hematuria, hypertension (from renin secretion), and fever may also be present. Anemia may occur in patients with subcapsular hemorrhage.
Treatment
Treatment follows a multimodality approach combining surgery, radiation therapy, and chemotherapy as defined by NWTS protocols, which have progressively improved outcomes. For unilateral resectable tumors, radical nephrectomy via a transabdominal incision is the procedure of choice. Retroperitoneal lymph node dissection is not standard, but regional lymph node biopsy is performed for staging. Avoiding tumor spillage during surgery is a major priority as spillage increases the risk of abdominal recurrence. For bilateral Wilms tumor (Stage V), preoperative chemotherapy followed by renal-sparing surgery is the preferred approach to preserve renal function. Wilms tumor is highly chemosensitive; actinomycin D and vincristine form the backbone of chemotherapy for favorable histology tumors, with doxorubicin added for higher-stage disease. Radiation therapy is used for Stage III and IV disease and for pulmonary metastases. Overall survival rates now exceed 85-90% for localized favorable histology disease, making Wilms tumor one of the success stories of pediatric oncology. - Smith and Tanagho's General Urology, 19e, p. 356-358
4. UROTHELIAL CARCINOMA OF THE BLADDER
Epidemiology and Overview
Urothelial (formerly called transitional cell) carcinoma is the most common malignancy of the urinary bladder and the predominant histological type, accounting for over 90% of bladder cancers. Bladder cancer is the fourth most common cancer in men and ninth in women in Western countries. It occurs most frequently in the sixth to eighth decades of life, with a male-to-female ratio of approximately 3:1. Smoking is the single most important risk factor, responsible for at least 50% of cases; chemical carcinogens (arylamines such as 2-naphthylamine and benzidine from dye, rubber, leather, and textile industries), analgesic abuse (phenacetin), cyclophosphamide exposure, pelvic radiation, and chronic bladder inflammation (especially Schistosoma haematobium infection, which is more strongly associated with squamous cell carcinoma) are other recognized risk factors.
Classification: Non-Muscle-Invasive vs. Muscle-Invasive
Urothelial carcinoma is fundamentally classified into non-muscle-invasive (superficial) disease and muscle-invasive disease, because this distinction drives treatment decisions entirely.
Non-muscle-invasive bladder cancer (NMIBC) comprises Ta (papillary tumor confined to urothelium), T1 (invasion into lamina propria), and Tis/CIS (flat, high-grade carcinoma in situ). About 70-75% of newly diagnosed bladder cancers are non-muscle-invasive. Ta tumors are often low-grade and papillary; Tis is flat, high-grade, and carries a high risk of progression to muscle-invasive disease. T1 tumors, by invading the lamina propria, have an intermediate risk. Importantly, CIS is a biologically aggressive lesion - it is flat (non-papillary), consists of malignant cells with high-grade nuclear features, and has a strong tendency to progress to invasive cancer.
Muscle-invasive bladder cancer (MIBC) includes T2 (invasion into muscularis propria), T3 (perivesical fat invasion), and T4 (invasion into adjacent structures). This group has a far worse prognosis and requires more aggressive therapy.
Molecular Pathogenesis
Two molecular pathways drive bladder tumorigenesis. The first - the low-grade papillary pathway - involves activating mutations in RAS or FGFR3 that lead to low-grade papillary tumors. These tumors are frequently recurrent but uncommonly progress to invasive disease. The second - the CIS/high-grade invasive pathway - involves loss of p53 and Rb function, leading to flat high-grade CIS that progresses to muscle-invasive carcinoma. Whole chromosome 9 deletions or monosomy 9 (loss of both CDKN2A/p16 loci on 9p and TSC1 on 9q) are among the earliest genetic events in bladder carcinogenesis.
Histologic Grading
Urothelial tumors are graded as low-grade or high-grade under the 2004 WHO classification, replacing the older Grade 1-3 system. Low-grade tumors have orderly urothelium with minimal nuclear atypia. High-grade tumors have disordered architecture, significant pleomorphism, frequent mitoses, and a higher propensity for invasion and metastasis. Flat CIS is, by definition, high-grade.
Histologic Variants
Several histologic variants of urothelial carcinoma exist, including micropapillary, plasmacytoid, sarcomatoid (most aggressive, with spindle cell morphology), nested, small cell, and squamous or glandular divergent differentiation. These variants carry prognostic and therapeutic implications.
Gross and Pathologic Features
Bladder carcinomas most commonly arise on the posterior wall and trigone. They may appear as papillary exophytic growths (more commonly low-grade) or as flat, velvety mucosal thickenings (CIS) or ulcerative, indurated lesions (invasive cancer). Multifocality is characteristic, with 30-40% of patients having more than one tumor at presentation, reflecting a "field effect" of carcinogen exposure on the entire urothelium (field cancerization). This same principle explains why patients treated for bladder CIS who undergo cystoprostatectomy commonly have urothelial carcinoma involving the prostatic ducts and acini (35-45% of cystoprostatectomy specimens). The urothelium of the upper urinary tract (ureters, renal pelvis) is similarly at risk - in 50% of renal pelvic urothelial tumors, there is a concurrent bladder tumor.
TNM Staging
- Ta: Non-invasive papillary carcinoma
- Tis: Carcinoma in situ (flat)
- T1: Invasion into subepithelial connective tissue (lamina propria)
- T2a/T2b: Invasion into superficial/deep muscularis propria
- T3a/T3b: Microscopic/macroscopic invasion into perivesical fat
- T4a: Invasion into prostate stroma, uterus, or vagina
- T4b: Invasion into pelvic or abdominal wall
- N1-3: Regional lymph node involvement
- M1: Distant metastasis
Clinical Presentation
The most common presenting symptom is painless gross hematuria, occurring in 85% of patients. Irritative voiding symptoms (frequency, urgency, dysuria) are particularly common with CIS. Obstructive symptoms, flank pain from ureteral obstruction, and pelvic pain occur in advanced disease. Systemic symptoms (weight loss, bone pain) suggest metastatic disease. Urinalysis showing microscopic hematuria in an at-risk patient warrants cystoscopic evaluation.
Diagnosis
Cystoscopy with biopsy (transurethral resection of bladder tumor, TURBT) is the gold standard for diagnosis and initial staging. Urine cytology is sensitive for high-grade disease and CIS but insensitive for low-grade tumors. Urine-based tumor markers (NMP22, BTA, FISH-based UroVysion) may complement cytology. CT urography (CTU) is performed to evaluate the upper urinary tract and assess for extravesical extension and lymph node involvement.
Treatment
Non-muscle-invasive disease: TURBT is both diagnostic and therapeutic for all NMIBC. Following complete TURBT, adjuvant intravesical therapy is given based on risk stratification. Low-risk tumors may receive a single immediate post-operative instillation of intravesical mitomycin C. Intermediate- and high-risk tumors receive a course of intravesical BCG (Bacillus Calmette-Guerin), the most effective intravesical agent, which reduces recurrence and progression. Maintenance BCG for 1-3 years is recommended for high-risk NMIBC. Re-TURBT is recommended within 6 weeks for T1 tumors or when the initial resection was incomplete, because residual tumor is found in 30-50% of cases. Patients who fail BCG therapy should be considered for radical cystectomy before progression to muscle-invasive disease.
Muscle-invasive disease: The standard of care is neoadjuvant cisplatin-based chemotherapy (MVAC - methotrexate, vinblastine, doxorubicin, cisplatin, or gemcitabine-cisplatin) followed by radical cystectomy. Neoadjuvant chemotherapy achieves complete pathologic response in 20-40% of patients and provides a survival advantage over cystectomy alone. Radical cystectomy entails removal of the bladder, perivesical fat, regional lymph nodes, and adjacent organs (prostate/seminal vesicles in men; uterus, cervix, and anterior vaginal wall in women), followed by urinary diversion. Carboplatin is significantly less efficacious than cisplatin in urothelial carcinoma and is not an adequate substitute even in cisplatin-ineligible patients, who should instead proceed directly to cystectomy or be enrolled in a clinical trial. For patients unwilling or unable to undergo cystectomy, bladder-preserving trimodality therapy - maximal TURBT followed by concurrent cisplatin-based chemoradiation - is an alternative. For patients with recurrence of muscle-invasive disease after chemoradiation, salvage radical cystectomy is performed in the absence of distant metastases.
Metastatic disease: Platinum-based chemotherapy (MVAC or gemcitabine-cisplatin) is first-line. Immune checkpoint inhibitors (pembrolizumab, atezolizumab) are approved for platinum-ineligible patients and as second-line therapy after platinum failure. Enfortumab vedotin (an antibody-drug conjugate targeting Nectin-4) and sacituzumab govitecan have shown significant activity. Erdafitinib is approved for tumors with FGFR3/2 alterations. The combination of enfortumab vedotin plus pembrolizumab has become a new standard of care in the first-line metastatic setting.
Adjuvant chemotherapy after radical cystectomy is used for pathologic T3/T4 or node-positive (pN+) disease in patients who did not receive neoadjuvant chemotherapy, though the evidence base is somewhat weaker than for neoadjuvant treatment. The standard approach is to prioritize neoadjuvant therapy; adjuvant cisplatin-based regimens are reasonable for those who did not receive it preoperatively. - Smith and Tanagho's General Urology, 19e, p. 969-980; Campbell-Walsh-Wein Urology
Summary Comparison Table
| Feature | RCC (Clear Cell) | Renal Oncocytoma | Nephroblastoma | Urothelial Ca (Bladder) |
|---|---|---|---|---|
| Cell of origin | Proximal tubule | Intercalated cells (collecting duct) | Metanephric blastema | Transitional epithelium |
| Age | 6th-7th decade | Any adult | Peak ~3 years | 6th-8th decade |
| Gender | M:F = 2:1 | Slight male predominance | Equal | M:F = 3:1 |
| Nature | Malignant | Benign | Malignant | Malignant |
| Key gene | VHL (3p25) | Complex I genes; chr 1/Y loss | WT1 (11p13) | p53, Rb, FGFR3 |
| Gross | Yellow, necrotic, vascular | Tan/mahogany, central scar | Large, multilobulated, gray | Papillary or flat/indurated |
| Classic symptom | Hematuria + flank pain + mass | Incidental | Abdominal mass | Painless hematuria |
| Spread | Renal vein, IVC | None (benign) | Renal vein, lungs, liver | Lymphatics, hematogenous |
| Key treatment | Partial/radical nephrectomy ± targeted therapy | Partial nephrectomy | Nephrectomy + chemo +/- RT | TURBT + BCG / Cystectomy + neoadjuvant chemo |
Sources: Robbins & Kumar Basic Pathology (9780323790185), Robbins Cotran & Kumar Pathologic Basis of Disease (9780443264528), Smith and Tanagho's General Urology 19e (9781259834332), Campbell-Walsh-Wein Urology (9780323546423), National Kidney Foundation Primer on Kidney Diseases 8e (9780323791229)
Tell me more in detail i'm not understanding
Asking for Preferences
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed. Here is a complete, from-scratch, student-friendly explanation of all four topics.
Complete Student Guide: Renal & Bladder Tumors - Explained From Scratch
HOW TO READ THIS GUIDE
Think of each tumor as a story with 5 chapters:
- Where does it come from? (Cell of origin)
- Why does it happen? (Pathogenesis/molecular mechanism - step by step)
- What does it look like? (Gross + histology)
- How does the patient present? (Clinical features)
- How do we treat it? (Management)
TOPIC 1: RENAL CELL CARCINOMA (RCC)
Step 1 - Where Does It Come From?
The kidney is made up of millions of tiny tubules that filter blood. RCC arises from the epithelial cells lining these renal tubules - specifically the proximal convoluted tubule (for most types) and the collecting duct intercalated cells (for chromophobe type). Since tubules are in the cortex, RCC is predominantly a cortical tumor.
It is the #1 malignant tumor of the kidney in adults, making up 80-85% of all primary renal malignancies. It accounts for about 2-3% of all adult cancers. Men get it twice as often as women, and it peaks in the 6th-7th decade of life.
Risk factors to memorize: Smoking (MOST important - doubles risk), obesity, hypertension, cadmium exposure, acquired polycystic disease from chronic dialysis (increases risk 30-fold), tuberous sclerosis.
Step 2 - Why Does It Happen? (Pathogenesis, explained simply)
There are 3 main subtypes of RCC, each with a different molecular mechanism. This is the part most students find confusing, so let's go very slowly.
A) CLEAR CELL RCC (most common - 65-80%)
The key gene: VHL (von Hippel-Lindau) on chromosome 3p25.
First, understand what VHL normally does. Under normal oxygen levels (normoxia), the VHL protein constantly destroys a molecule called HIF-1 (Hypoxia Inducible Factor). Think of VHL as a "garbage collector" that keeps HIF-1 levels low when oxygen is plentiful.
HIF-1 is a transcription factor that, when active, turns on genes that help cells survive with low oxygen - it tells cells to grow new blood vessels (by making VEGF) and to proliferate. Under normal conditions, this is switched off by VHL.
Now what happens in Clear Cell RCC:
- Both copies of the VHL gene are lost/mutated (two-hit model: chromosome 3p deletion knocks out one copy; somatic mutation/hypermethylation knocks out the second copy).
- With no VHL "garbage collector" functioning, HIF-1 is never destroyed.
- HIF-1 accumulates even with normal oxygen → it acts as if the cell is hypoxic all the time.
- This causes constant overexpression of VEGF (→ rich blood vessel formation in the tumor, explaining its high vascularity) and IGF-1 (→ cell proliferation).
- HIF-1 also teams up with MYC to rewire cell metabolism to favor growth.
- Additionally, mutations in histone-methylation genes alter the epigenome, contributing further.
Result: A richly vascularized, rapidly growing tumor.
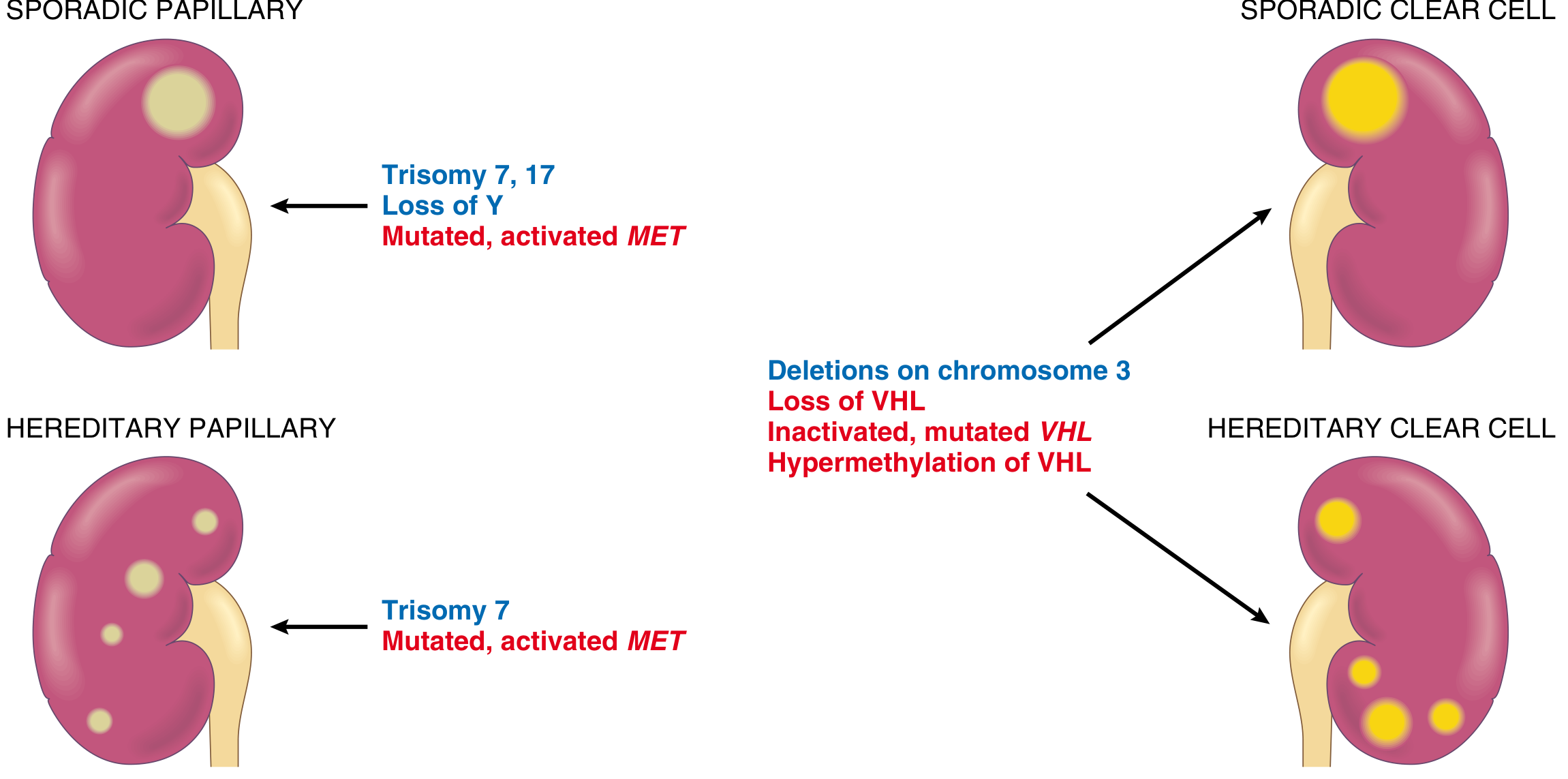
Cytogenetics (blue) and genetics (red) of clear cell versus papillary renal cell carcinoma. - Robbins Cotran & Kumar Pathologic Basis of Disease
Familial connection - VHL Disease: This is an autosomal dominant syndrome where patients are born with one already-mutated copy of VHL. A single additional somatic hit in the kidney cell causes bilateral, multiple clear cell RCCs in 40-60% of these patients. They also get hemangioblastomas (benign blood vessel tumors) of the cerebellum and retina.
B) PAPILLARY RCC (10-15%)
The key gene: MET proto-oncogene on chromosome 7q.
MET encodes the receptor for hepatocyte growth factor (HGF/scatter factor). Normally, MET is activated only when HGF binds to it, telling cells to grow, migrate, and differentiate in a controlled way.
In Papillary RCC:
- Gain-of-function mutations or extra copies of MET (from trisomies of chromosome 7 and 17) cause the MET receptor to be permanently overactive - it fires growth signals non-stop, even without HGF.
- This drives abnormal proliferation of proximal/distal tubular cells.
- Since the mutation can be in every kidney cell (germline/hereditary form) or in many cells (sporadic, from trisomy 7), these tumors are often multifocal and bilateral.
C) CHROMOPHOBE RCC (5%)
Cell of origin: Intercalated cells of the collecting duct.
The molecular mechanism is less well-understood but involves multiple losses of entire chromosomes - leading to extreme hypoploidy (very few chromosomes). This is the subtype with the best prognosis among the three.
Step 3 - What Does It Look Like?
Gross Appearance (what you see with naked eye)
Clear cell RCC is typically a large, solitary, spherical mass (3-15 cm) in the cortex. When you cut it open, the surface is bright yellow-to-orange - this color is caused by the massive amount of lipid (fat) and glycogen stuffed inside the tumor cells. There are areas of necrosis (dead, grayish tissue), hemorrhage (red), and cystic change.
The most clinically important gross feature is renal vein invasion - the tumor physically grows into the renal vein like a snake, can extend all the way up the inferior vena cava (IVC), and in extreme cases reaches the right side of the heart. This is a classic exam point.
Cross-section of a kidney showing a yellow RCC (asterisk) with tumor thrombus in the renal vein (arrow). - Robbins & Kumar Basic Pathology
Histology (what you see under microscope)
Clear cell type: Cells are arranged in nests or alveoli (small clusters) separated by delicate, branching blood vessels (fibrovascular stroma). The cells have clear, vacuolated cytoplasm (the lipid and glycogen was washed out during processing, leaving empty-looking cells) with distinct cell membranes and small, round nuclei. The rich vascularity is visible between nests.
Papillary type: Cells form finger-like projections (papillae) with a fibrovascular core. A classic feature is foamy macrophages (lipid-laden macrophages) in the stalks of the papillae. Cells are cuboidal to low columnar with eosinophilic to clear cytoplasm. Psammoma bodies (concentric calcifications) may be seen.
Chromophobe type: Cells are large with pale eosinophilic cytoplasm, very prominent cell membranes (you can see each cell border clearly), and often have a perinuclear halo (clear zone around the nucleus). Cells concentrate around blood vessels. Arranged in solid sheets.
RCC histological subtypes. (A) Clear cell - empty vacuolated cells in nests. (B) Papillary - papillary fronds with foamy macrophages. (C) Chromophobe - eosinophilic cells with prominent membranes. - Robbins & Kumar Basic Pathology
Step 4 - How Does the Patient Present?
Classic triad (only seen in 10% of patients): Painless hematuria + flank pain + palpable abdominal/flank mass.
Most common single symptom: Painless gross hematuria (blood in urine, in over 50% of cases). Hematuria is intermittent - it comes and goes, so patients may ignore it. This is dangerous.
Today: Most RCCs are found incidentally on CT/ultrasound done for another reason (e.g., for abdominal pain or stones).
Paraneoplastic syndromes - RCC is famous for producing hormones it shouldn't, causing distant effects:
- Polycythemia (high RBC count) - tumor secretes erythropoietin - seen in 5-10%
- Hypercalcemia - tumor secretes PTHrP
- Hypertension - tumor secretes renin
- Cushing syndrome - ectopic ACTH
- Hepatic dysfunction without liver metastasis (Stauffer syndrome) - rare
- Fever, weight loss, fatigue (constitutional symptoms)
Metastases pattern: RCC is the "great mimic" - it metastasizes widely before causing local symptoms. Most common sites: Lungs (>50%), Bones (33%), then lymph nodes, liver, adrenal, brain.
Step 5 - How Do We Treat It?
Localized disease:
- Partial nephrectomy (remove only the tumor, preserve rest of kidney) - preferred for tumors <4 cm (T1a) and when feasible for larger tumors.
- Radical nephrectomy (remove entire kidney + fat + adrenal) for larger/complex tumors.
- Even with renal vein/IVC thrombus - surgery can still be curative!
Metastatic disease: RCC does NOT respond well to standard chemotherapy. Instead:
- Anti-VEGF/anti-angiogenic drugs (sunitinib, pazopanib, axitinib) - block the VEGF pathway driven by HIF overactivation.
- mTOR inhibitors (everolimus, temsirolimus)
- Immune checkpoint inhibitors (nivolumab + ipilimumab combination, pembrolizumab) - now first-line for many metastatic patients.
Prognosis:
- No metastases → ~100% 5-year survival
- Renal vein invasion or perinephric fat involvement → ~60% 5-year survival
- Overall average → ~70% 5-year survival
TOPIC 2: RENAL ONCOCYTOMA
The Big Picture First
Oncocytoma is a benign tumor - it does NOT metastasize and does NOT kill the patient. But it is important because it looks worryingly similar to malignant RCC on imaging, so patients often end up being operated on to confirm the diagnosis.
Cell of Origin
Arises from the intercalated (A) cells of the collecting duct - the same origin as chromophobe RCC, which is why the two are so difficult to distinguish. It represents 3-7% of all renal neoplasms.
Why Does It Happen? (Pathogenesis)
The central event is a loss of Complex I of the mitochondrial electron transport chain (the chain responsible for making ATP by oxidative phosphorylation). When Complex I is lost, the cell cannot perform normal oxidative phosphorylation. This triggers a compensatory feedback loop that says "make more mitochondria to compensate." The cell responds by massively proliferating mitochondria. The tumor cell ends up with its cytoplasm crammed full of mitochondria. This explains everything about this tumor:
- Cytoplasm is full of mitochondria → appears bright pink/eosinophilic (granular) under the microscope (mitochondria take up eosin stain).
- The massive mitochondrial content makes the tumor brownish-tan/mahogany in color to the naked eye (mitochondria give it that color).
- On electron microscopy, you can literally see the cytoplasm packed densely with mitochondria.
Chromosomal abnormalities: Loss of chromosomes 1 and Y, rearrangements of the cyclin D1 locus.
Familial cases (oncocytosis): Multiple bilateral oncocytomas, seen in tuberous sclerosis patients.
Gross Appearance
- Tan or mahogany brown color (not yellow like clear cell RCC)
- Well-circumscribed, with a fibrous capsule
- Central stellate scar - a characteristic central scar in a star/spoke shape - present in about 1/3 of cases. This scar can sometimes be seen on CT/MRI and suggests oncocytoma, but is NOT diagnostic.
- Can grow very large - up to 12 cm.
Histology
Uniform, round cells with abundant granular eosinophilic cytoplasm and small, round, benign-appearing nuclei with small but conspicuous nucleoli. Arranged in nests and tubules within a loose stroma. No significant nuclear atypia, no necrosis, no mitotic figures - these benign features distinguish it from RCC.
On electron microscopy: cytoplasm is packed with mitochondria (this is the definitive ultrastructural finding).
Renal oncocytoma. (A) Uniform cells with granular eosinophilic cytoplasm. (B) Electron micrograph showing cytoplasm densely packed with mitochondria. - Robbins & Kumar Basic Pathology
Clinical Features and Why It's Tricky
Most oncocytomas are found incidentally on imaging. They cause no symptoms unless very large. The problem is that chromophobe RCC (a malignant tumor) looks almost identical on imaging AND on regular histology. Even experienced radiologists and pathologists sometimes cannot tell them apart without special studies. Because you cannot safely exclude malignancy preoperatively, most patients undergo partial nephrectomy. The tumor is then confirmed benign after surgery.
Important warning: In 10-30% of patients with multiple oncocytic nodules (oncocytosis), a concurrent RCC is also present. So follow-up imaging is needed even after diagnosing oncocytoma.
Treatment
Partial nephrectomy is curative. No chemotherapy, no radiation, no further treatment needed.
TOPIC 3: NEPHROBLASTOMA (WILMS TUMOR)
The Big Picture First
Wilms tumor is the kidney version of "a tumor that arises because the kidney failed to develop normally." It is essentially fetal kidney tissue that kept growing instead of maturing. It is the #1 primary renal malignancy of childhood (most common solid renal tumor in children), peaking at age 3 years. It is extremely chemosensitive and has an excellent cure rate (>85%).
Cell of Origin and Concept
During fetal kidney development, the metanephric blastema (a mass of primitive embryonic cells) differentiates into the various components of the mature kidney. If some of these primitive cells fail to differentiate and persist after birth, they are called nephrogenic rests - these are the precursors to Wilms tumor.
Think of it this way: the kidney is supposed to "graduate" all its cells from primitive blastema to mature tubules and glomeruli. Wilms tumor happens when some cells "refuse to graduate" and instead start dividing abnormally.
Pathogenesis - The Genetics (Step by Step)
The Two-Hit Hypothesis (Knudson, 1972)
This is the same concept as retinoblastoma. You need two mutations in the same cell to develop Wilms tumor because the relevant genes are tumor suppressors (both copies must be lost to remove the brake on cell growth).
- Sporadic form: Both mutations happen after birth, in the same kidney cell (post-zygotic). These patients typically present later, with unilateral tumor.
- Familial/hereditary form: The first mutation is present in every cell from birth (inherited germline mutation). Only ONE more somatic mutation is needed in any kidney cell to start a tumor. This explains why familial cases present EARLIER in life and are more likely to be BILATERAL and MULTIFOCAL.
Key Gene: WT1 (Chromosome 11p13)
WT1 (Wilms Tumor gene 1) encodes a zinc-finger transcription factor essential for normal kidney and gonadal development. Loss of both WT1 copies removes a critical brake on proliferation of metanephric blastema cells, allowing them to keep dividing. However, WT1 mutations are found in only 5-10% of sporadic Wilms tumors, meaning other genes (WT2 at 11p15, which involves imprinting of IGF-1, H19, p57) also play a role.
Associated Syndromes (Very High-Yield for Exams)
| Syndrome | Features | Gene |
|---|---|---|
| WAGR | Wilms + Aniridia + Genitourinary malformations + Retardation | Deletion 11p13 (includes WT1 and PAX6) |
| Beckwith-Wiedemann | Macroglossia + macrosomia + organomegaly + hemihypertrophy + ear creases | 11p15 (IGF-1, H19, p57 imprinting) |
| Denys-Drash | Gonadal dysgenesis + nephropathy + Wilms tumor | WT1 point mutation |
Gross Appearance
Wilms tumors are typically large, multilobulated masses that appear grey or tan, with focal areas of hemorrhage (red-brown) and necrosis (pale/yellow). They can be enormous - filling the entire abdomen. A fibrous pseudocapsule sometimes surrounds them. They disseminate by direct extension through the renal capsule, into the renal vein/IVC, and via lymphatics.
Histology - The Triphasic Pattern (The Key to Recognizing Wilms on Biopsy)
The classic Wilms tumor has THREE components in varying proportions - this is what gives it away under the microscope:
-
Blastema - Primitive small round blue cells, tightly packed, with hyperchromatic nuclei and scant cytoplasm. This represents the undifferentiated embryonic kidney cells that failed to mature. This is the most cellular-looking and "blue" component.
-
Epithelium - Tubular and glomeruloid structures attempting to form kidney-like units. You see primitive tubules and structures resembling early glomeruli. This is the differentiating part.
-
Stroma - Loose spindle cells, often with smooth or skeletal muscle differentiation. This is the supportive/connective tissue component.
Wilms tumor with characteristic tubular/glomeruloid structures and blastema. - Smith and Tanagho's General Urology
Histologic Grading: Favorable vs. Unfavorable
This is clinically very important because it changes the treatment intensity.
Favorable histology (good prognosis): No anaplasia. Standard triphasic Wilms tumor.
Unfavorable histology (bad prognosis): Contains anaplasia (extreme nuclear enlargement, hyperchromasia, and abnormal mitotic figures). Anaplasia = p53 mutation, occurs in 5% of Wilms tumors, more in older children and African-Americans.
Two tumors that look like Wilms but are NOT and are far more aggressive: Clear cell sarcoma of the kidney (metastasizes to bone - "bone-metastasizing renal tumor of childhood") and Rhabdoid tumor of the kidney (most aggressive, often fatal).
Clinical Features
- Classic presentation: A parent notices a smooth, large, non-tender abdominal mass in a 3-year-old child while bathing them. This is the textbook scenario.
- Hypertension from renin secretion by the tumor.
- Hematuria (microscopic or gross).
- Fever.
- Abdominal pain is less common.
Metastases at diagnosis in 10-15% of cases - most common: lungs (85-95%), then liver.
NWTS Staging
- Stage I: Tumor entirely within kidney, completely removed.
- Stage II: Beyond kidney but completely removed (capsule breach, local spillage).
- Stage III: Residual disease in abdomen (positive nodes, peritoneal spill, incomplete resection).
- Stage IV: Hematogenous metastases (lung, liver, bone, brain).
- Stage V: Bilateral kidney involvement.
Treatment (Multimodality - Surgery + Chemo + RT)
Surgery: Radical nephrectomy is the standard for unilateral disease. The number one operative priority is avoiding tumor spillage - spillage turns a Stage I/II tumor into Stage III and increases abdominal recurrence. For bilateral (Stage V) disease, give preoperative chemotherapy first to shrink tumors, then do nephron-sparing surgery to save kidney function.
Chemotherapy: Wilms tumor is remarkably chemosensitive. The backbone drugs are:
- Stages I-III favorable histology: Actinomycin D + Vincristine
- Stage IV or unfavorable histology: Actinomycin D + Vincristine + Doxorubicin
Radiation: For Stage III and IV disease (abdominal/whole-lung irradiation for pulmonary mets).
Prognosis: Excellent - overall 5-year survival >85-90%. Even Stage IV favorable histology has good cure rates with aggressive multimodality treatment. Anaplastic (unfavorable) histology has a worse prognosis.
TOPIC 4: UROTHELIAL CARCINOMA OF THE BLADDER
The Big Picture First
The bladder is lined by a special epithelium called urothelium (also called transitional epithelium) - this is a unique epithelium that can stretch when the bladder fills and shrink when empty. Cancer arising from these cells is called urothelial carcinoma (previously called transitional cell carcinoma/TCC). It is the most common bladder cancer (>90% of bladder malignancies).
Think of the urothelium as a stretch-resistant lining constantly exposed to urine and anything dissolved in it. Carcinogens in urine directly contact the urothelium for hours every day - this is why the bladder is so susceptible to carcinogen-driven cancer.
Epidemiology
- 4th most common cancer in men, 9th in women (in Western countries).
- Male:Female = 3:1
- Peak: 6th-8th decade.
- #1 risk factor: Cigarette smoking (responsible for ~50% of cases). Carcinogens in tobacco are excreted in urine, bathing the urothelium.
- Occupational exposure to arylamines (2-naphthylamine, benzidine) - dye workers, rubber workers, leather workers. These chemicals are also excreted in urine and directly contact the urothelium.
- Cyclophosphamide (immunosuppressant/chemotherapy) - its metabolite acrolein is directly toxic to the urothelium.
- Pelvic radiation.
- Schistosoma haematobium infection (leads more to squamous cell carcinoma, not urothelial).
Why Does It Happen? Two Molecular Pathways
This is essential to understand because it explains why there are two completely different types of bladder cancer with different behaviors.
Pathway 1 - Low-grade papillary (superficial) pathway:
RAS or FGFR3 activating mutations → abnormal cell signaling → cells proliferate into finger-like papillary projections into the bladder lumen → they don't normally invade the muscle wall. These tumors are frequently recurrent (come back again and again) but rarely progress to become invasive and life-threatening. Think of them as "annoying but not usually deadly."
Pathway 2 - High-grade/invasive/CIS pathway:
Loss of p53 (chromosome 17p) and Rb (chromosome 13q) tumor suppressor genes, combined with loss of chromosome 9 (both 9p and 9q) → cells with severe nuclear atypia proliferate → flat Carcinoma In Situ (CIS) → progress to muscle-invasive cancer. This pathway leads to aggressive tumors that invade the bladder wall deeply and metastasize. Chromosome 9 deletions (monosomy 9) are among the earliest events in all bladder carcinogenesis.
Classification: The Most Important Clinical Distinction
Non-Muscle-Invasive Bladder Cancer (NMIBC) = tumor has NOT yet broken through the muscularis propria (the thick muscle wall). This is 70-75% of newly diagnosed cases.
- Ta: Papillary tumor confined to the urothelium only (hasn't even invaded the lamina propria)
- Tis (CIS): Flat, high-grade carcinoma in situ - cells look malignant but haven't invaded yet
- T1: Invades into the lamina propria (the layer below the urothelium) but not the muscle
Muscle-Invasive Bladder Cancer (MIBC) = tumor has broken into and through the muscularis propria. This is far more dangerous.
- T2: Into the muscle (T2a = superficial muscle, T2b = deep muscle)
- T3: Through the muscle into perivesical fat
- T4: Into adjacent organs (prostate, uterus, vagina, pelvic wall)
Why this distinction matters so much: NMIBC is treated with endoscopic surgery + intravesical therapy (drugs put directly into the bladder). MIBC requires major surgery (removal of the entire bladder) or chemoradiation. The treatment is completely different.
Gross Appearance
Papillary tumors (usually low-grade, Ta/T1): Frond-like, finger-like projections growing into the bladder lumen, resembling a sea anemone or cauliflower. They are fragile and bleed easily, which is why they cause hematuria.
Flat CIS: You cannot see it with the naked eye - the bladder mucosa looks normal or slightly red and velvety. It is invisible to the surgeon's eye and is diagnosed by random biopsies and urine cytology showing malignant cells.
Invasive tumors (T2-T4): Solid, indurated (hardened), ulcerated masses embedded in the bladder wall. When you cut the bladder wall, you can see the tumor infiltrating into and through the muscle.
Urothelial carcinoma of the renal pelvis - the pelvis opened to show the nodular irregular neoplasm. - Robbins Cotran & Kumar Pathologic Basis of Disease
Histology
The urothelium normally is 3-7 cells thick with superficial "umbrella cells." In low-grade urothelial carcinoma, you see papillary fronds lined by urothelium with mild nuclear enlargement and maintained cell polarity. In high-grade carcinoma, the lining cells show marked nuclear pleomorphism, loss of polarity, prominent nucleoli, and increased mitoses. In CIS, the full thickness of the urothelium is replaced by markedly atypical cells without invasion.
When the tumor invades the lamina propria (T1), you see irregular nests of atypical urothelial cells breaking through the basement membrane into the connective tissue. When it invades muscle (T2), nests of tumor cells are seen within the smooth muscle bundles of the muscularis propria.
Clinical Features
Painless gross hematuria - the single most common and important symptom. It is intermittent (comes and goes), which leads patients to ignore it. Any adult with painless hematuria should be suspected of bladder cancer until proven otherwise.
Irritative voiding symptoms (urgency, frequency, dysuria) - particularly associated with CIS, because the entire bladder mucosa is irritated.
Obstructive symptoms (poor stream, retention) - if the tumor is near the bladder neck or urethral orifice.
Unilateral flank pain/hydronephrosis - if the tumor obstructs a ureteral orifice.
Advanced disease: Pelvic pain, weight loss, bone pain (from metastases).
Field cancerization concept: Because the ENTIRE urothelium of the urinary tract (from renal pelvis to urethra) was exposed to the same carcinogens, multiple tumors can develop anywhere in the system simultaneously. In 50% of renal pelvic urothelial tumors, there is a concurrent bladder tumor. Likewise, 35-45% of bladder cystoprostatectomy specimens contain tumor in the prostatic ducts/acini. Recurrence in the bladder after treatment is extremely common - this is why lifelong cystoscopic surveillance is essential.
Diagnosis
- Cystoscopy + TURBT (Transurethral Resection of Bladder Tumor): Gold standard. The urologist passes a camera into the bladder, visualizes the tumor, and removes it endoscopically. This is both diagnostic and therapeutic for NMIBC.
- Urine cytology: Sensitive for high-grade disease and CIS (malignant-looking cells shed into urine). Less useful for low-grade tumors.
- CT urography: To evaluate the entire upper urinary tract (ureters, renal pelvis) and detect extravesical spread, lymph node involvement.
Treatment
Non-Muscle-Invasive Disease (NMIBC)
- Step 1: TURBT - Remove all visible tumor endoscopically.
- Step 2: Immediate post-operative intravesical mitomycin C (a single instillation into the bladder right after TURBT) - for low-risk patients to reduce recurrence.
- Step 3: BCG (Bacillus Calmette-Guerin) intravesical therapy - for intermediate/high-risk NMIBC (T1 tumors, CIS, high-grade Ta). BCG is a bacterial immunotherapy instilled directly into the bladder; it triggers an immune response that kills residual tumor cells and prevents recurrence. This is the most effective intravesical treatment. Maintenance BCG is given for 1-3 years.
- Re-TURBT: A second TURBT 6 weeks later is done for T1 tumors - because 30-50% of these have residual tumor left behind after the first resection.
- Patients who fail BCG: Should undergo radical cystectomy to prevent progression to muscle-invasive disease.
Muscle-Invasive Disease (MIBC)
- Neoadjuvant cisplatin-based chemotherapy FIRST - then surgery. This is the standard of care. The regimen is MVAC (Methotrexate + Vinblastine + doxorubicin/Adriamycin + Cisplatin) or gemcitabine + cisplatin. Given before surgery to eliminate micrometastatic disease and shrink the primary tumor. Complete pathologic response in 20-40% of patients. Provides a survival advantage over surgery alone.
- Radical cystectomy: Remove the entire bladder + perivesical fat + regional lymph nodes. In men, also remove prostate and seminal vesicles. In women, also remove uterus, cervix, and anterior vaginal wall. Then urinary diversion is needed (ileal conduit/neobladder).
- Important: Carboplatin is NOT a substitute for cisplatin in bladder cancer even if the patient cannot tolerate cisplatin - it is significantly less effective. Patients who cannot receive cisplatin should go straight to cystectomy without neoadjuvant chemotherapy.
- Bladder-preserving trimodality therapy (for those who refuse/cannot have cystectomy): Maximum TURBT + concurrent chemoradiation (cisplatin + radiation). Approximately 30-50% can achieve long-term disease-free survival with intact bladder. Contraindicated if CIS, multifocal disease, or hydronephrosis is present.
Metastatic Disease
- First-line: MVAC or gemcitabine + cisplatin
- Second-line: Immune checkpoint inhibitors (pembrolizumab, atezolizumab)
- Novel agents: Enfortumab vedotin (antibody-drug conjugate) + pembrolizumab combination is now a major first-line option in metastatic disease.
- FGFR3 mutations: Erdafitinib (FGFR inhibitor) is approved for FGFR2/3-altered tumors.
MASTER COMPARISON - All 4 Tumors at a Glance
| RCC | Oncocytoma | Nephroblastoma | Urothelial Ca | |
|---|---|---|---|---|
| Nature | Malignant | Benign | Malignant | Malignant |
| Who gets it | Adults, 60s, M>F | Adults | Children, age 3, M=F | Adults, 60-70s, M>F 3:1 |
| Cell of origin | Proximal tubule epithelium | Intercalated cells of collecting duct | Metanephric blastema | Urothelial epithelium |
| Location | Renal cortex | Renal cortex | Anywhere in kidney | Bladder mucosa |
| Key gene / molecular change | VHL loss (clear cell), MET gain (papillary), Chr losses (chromophobe) | Complex I loss → mitochondrial proliferation | WT1 (11p13) loss | p53/Rb loss (invasive), FGFR3/RAS gain (papillary) |
| Gross color | Yellow-orange (lipid-rich) | Tan/mahogany brown (mitochondria-rich) | Grey-tan with hemorrhage/necrosis | Papillary/pink OR ulcerated |
| Classic histology | Nests of clear cells + delicate vasculature | Uniform pink granular cells + central scar | TRIPHASIC: blastema + epithelium + stroma | Papillary fronds OR flat CIS |
| #1 symptom | Painless hematuria | Incidental finding | Abdominal mass (parent finds it) | Painless intermittent gross hematuria |
| Famous spread | Renal vein → IVC → right heart | Does NOT spread (benign) | Lungs (85-95%), then liver | Superficial recurrence; → muscle invasion → metastasis |
| Treatment | Partial/radical nephrectomy ± TKI/immunotherapy | Partial nephrectomy (curative) | Nephrectomy + actinomycin D/vincristine ± RT | TURBT + BCG (NMIBC) / Neoadjuvant chemo + cystectomy (MIBC) |
| Prognosis | ~70% 5-yr overall | Excellent (benign) | >85% 5-yr overall | Variable: NMIBC good; MIBC ~50% 5-yr |
Sources: Robbins & Kumar Basic Pathology (9780323790185, p.531-534), Robbins Cotran & Kumar Pathologic Basis of Disease (9780443264528, p.878-882), Smith and Tanagho's General Urology 19e (9781259834332, p.356-358, 424-426), Campbell-Walsh-Wein Urology (9780323546423)
This is a shared conversation. Sign in to Orris to start your own chat.