Myocardial infarction. Pathogenesis. Classification.
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, authoritative content from Robbins Pathologic Basis of Disease. Let me compile the full answer.
Myocardial Infarction: Pathogenesis and Classification
Definition
Myocardial infarction (MI) is the death of cardiac muscle due to prolonged ischemia. Roughly 800,000 MIs occur annually in the United States. The overwhelming majority are caused by atherosclerosis of the coronary arteries. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
Pathogenesis
1. Atherosclerotic Background
Nearly all MIs arise on a substrate of coronary atherosclerosis. Atheromatous plaques progressively narrow the lumen and create a vulnerable substrate for acute events. Importantly, the culprit plaque often does not produce a critical (>70%) stenosis beforehand - it is the acute change in the plaque, not the degree of chronic stenosis, that precipitates infarction.
2. Acute Plaque Disruption - the Central Event
The typical sequence of events:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to blood.
- Platelets adhere, aggregate, and activate, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Activation of the coagulation cascade by tissue factor and other mechanisms adds to the growing thrombus.
- Within minutes, the thrombus can completely occlude the coronary artery lumen.
Angiography within 4 hours of MI onset demonstrates coronary thrombosis in ~90% of cases. By 12-24 hours (without intervention), only ~60% show thrombosis, because some occlusions clear spontaneously.
3. Non-Atherothrombotic Mechanisms (~10% of Cases)
- Vasospasm - with or without atherosclerosis; may be triggered by cocaine, ephedrine, or intrinsic mechanisms
- Embolism - from mural thrombus (AF), infective endocarditis vegetations, prosthetic material, or paradoxical emboli via patent foramen ovale
- Vasculitis - small intramural vessel inflammation
- Hematologic - sickle cell disease, hypercoagulable states
- Amyloid deposition in vascular walls
4. Myocardial Response to Ischemia
The outcome depends on severity and duration of blood flow deprivation:
| Event | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
Source: Robbins, Cotran & Kumar - Table 12.4
The first biochemical consequence is cessation of aerobic metabolism within seconds → inadequate high-energy phosphate production (ATP, creatine phosphate) + accumulation of lactic acid. Contractility ceases within ~1 minute. Ultrastructural changes (myofibrillar relaxation, glycogen depletion, mitochondrial swelling) are initially reversible. Only ischemia lasting 20-40 minutes with flow ≤10% of normal produces irreversible necrosis.
5. Wavefront Phenomenon
Irreversible injury begins in the subendocardial zone - this region is most vulnerable because:
- It is the last to receive blood from epicardial vessels
- It is exposed to higher intramural pressures that impede inflow
With prolonged ischemia, a wavefront of cell death moves centripetally (outward toward the epicardium). A full-thickness (transmural) infarct evolves over 6-12 hours.
6. Reperfusion Injury
Restoration of blood flow salvages ischemic but still-viable myocardium - but can also cause reperfusion injury accounting for up to 50% of final infarct size. Mechanisms include:
- Mitochondrial dysfunction - altered permeability, outer membrane rupture, apoptosis
- Myocyte hypercontracture - calcium overload causes uncontrolled actin-myosin interaction → cytoskeletal damage
- Free radical generation - O2•⁻, H2O2, •OH, peroxynitrite damage membrane proteins and phospholipids within minutes of reperfusion
- Leukocyte aggregation - microvascular plugging causing "no-reflow" phenomenon; leukocytes also release proteases/elastases
- Platelet and complement activation - endothelial injury
Reperfused infarcts are typically hemorrhagic grossly. Microscopically, irreversibly injured myocytes show contraction band necrosis (intense eosinophilic bands of hypercontracted sarcomeres from calcium influx).
Special states:
- Stunned myocardium - prolonged contractile dysfunction after short-term ischemia, recovers over days
- Hibernating myocardium - chronically reduced metabolism/function from chronic sublethal ischemia; recovers with revascularization
Morphological Evolution (Classification by Time)
The gross and histologic appearance evolves predictably (Table 12.5 from Robbins):
| Time | Gross Features | Light Microscopy | Electron Microscopy |
|---|---|---|---|
| 0-0.5 hr (reversible) | None | None | Myofibrillar relaxation, glycogen loss, mitochondrial swelling |
| 0.5-4 hr | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Dark mottling (occasional) | Early coagulative necrosis; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Coagulative necrosis; nuclear pyknosis; myocyte hypereosinophilia; contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan center | Coagulative necrosis with loss of nuclei and striations; brisk neutrophilic infiltrate | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Disintegrating dead myofibers; dying neutrophils; macrophage phagocytosis at border; early granulation tissue | - |
| 7-10 days | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed granulation tissue with prominent vascularization at margins | - |
| 10-14 days | Red-tan margins; yellow-tan center | Active granulation tissue and early collagen deposition | - |
| 2-8 weeks | Gray-white fibrous scar forming | Increased collagen deposition with decreased cellularity | - |
| >2 months | Complete fibrous scar (white) | Dense collagen scar | - |
Classification
I. By Depth of Necrosis (Anatomic / Pathological)
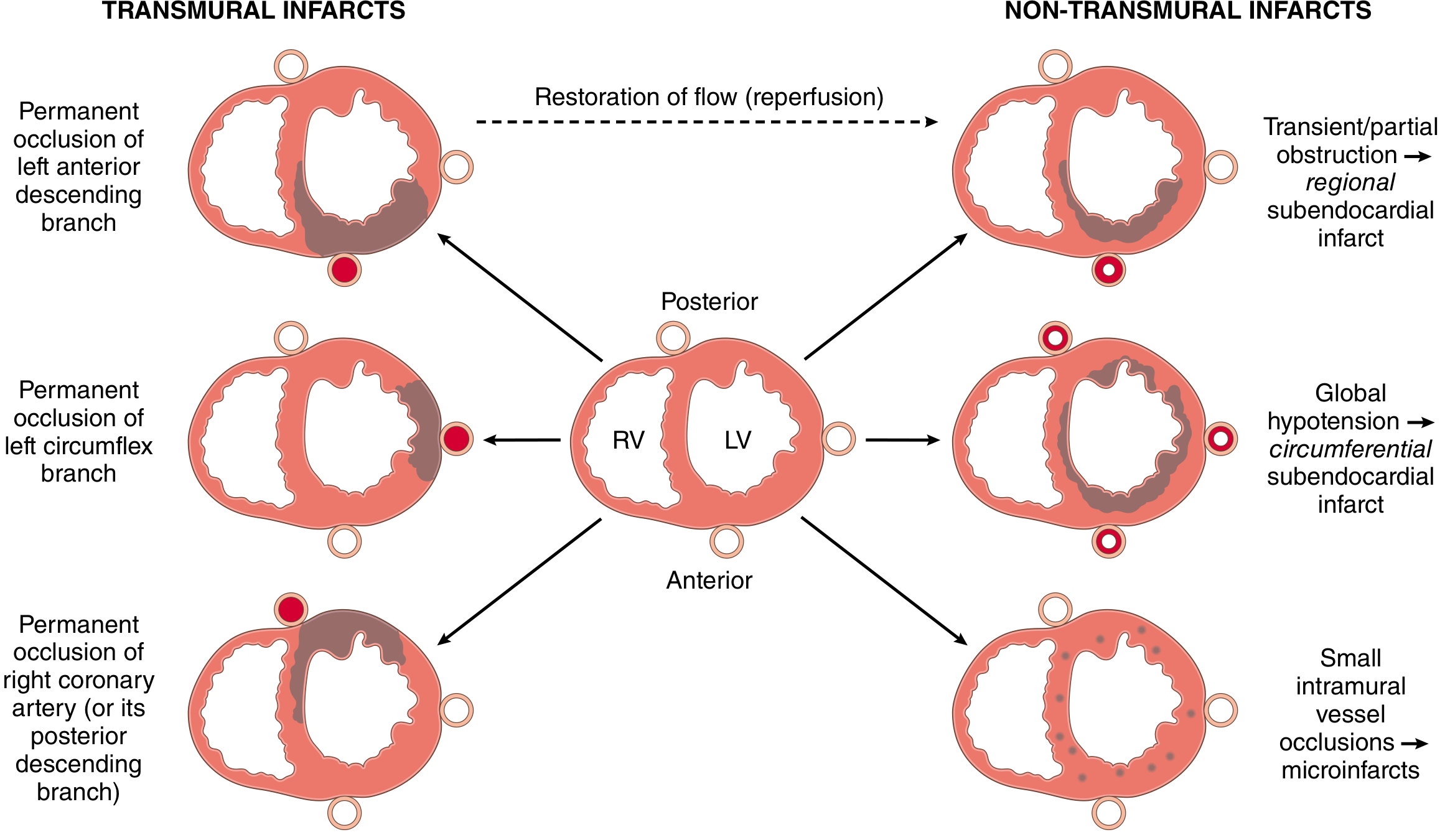
1. Transmural MI
- Necrosis involves the full thickness (or near-full thickness) of the ventricular wall
- Results from permanent epicardial vessel occlusion (atherothrombosis)
- Associated with STEMI on ECG
- A narrow rim (~0.1 mm) immediately beneath the endocardium is preserved by diffusion from the ventricular lumen
2. Subendocardial (Non-transmural) MI
- Necrosis limited to the inner one-third to half of the ventricular wall
- Results from:
- Transient/partial coronary obstruction (with reperfusion)
- Severe global hypoperfusion (circumferential subendocardial infarct)
- Associated with NSTEMI on ECG
- Types:
- Regional subendocardial - in the territory of a partially obstructed artery
- Circumferential subendocardial - affects the entire inner ring of the LV (from global hypotension/shock)
3. Microinfarction
- Multiple tiny foci from small intramural vessel pathology (microembolism, vasculitis, catecholamine-induced vasospasm)
II. By Coronary Artery Territory (Localization)
| Coronary Artery | Frequency | Territory Infarcted |
|---|---|---|
| LAD | 40-50% | Anterior LV wall near apex; anterior 2/3 of ventricular septum; apex circumferentially |
| RCA | 30-40% | Inferior/posterior LV wall; posterior 1/3 of septum; inferior/posterior RV free wall |
| LCX | 15-20% | Lateral LV wall (except apex) |
III. By ECG Pattern (Clinical)
| Type | ECG | Pathology |
|---|---|---|
| STEMI (ST-elevation MI) | ST elevation → Q waves | Full-thickness (transmural) occlusion; complete coronary occlusion |
| NSTEMI (Non-ST-elevation MI) | ST depression, T-wave changes; no Q waves | Subendocardial or partial-thickness; troponin elevated without ST elevation |
| Unstable Angina | Transient ECG changes; no troponin rise | No necrosis; mural thrombus without complete occlusion |
IV. By Cause - The Fourth Universal Definition (Types 1-5)
The Universal Definition of Myocardial Infarction (4th edition, 2018) classifies MI into 5 types based on mechanism:
| Type | Name | Mechanism |
|---|---|---|
| Type 1 | Spontaneous MI | Atherosclerotic plaque disruption/erosion with coronary thrombosis |
| Type 2 | MI due to ischemic imbalance | Supply-demand mismatch without plaque rupture (e.g., coronary spasm, anemia, hypotension, tachyarrhythmia, hypertensive crisis) |
| Type 3 | MI causing sudden cardiac death | Death before biomarkers can be obtained; ECG evidence of ischemia |
| Type 4a | PCI-related MI | MI within 48 hours of percutaneous coronary intervention |
| Type 4b | Stent thrombosis | Angiographically confirmed stent/scaffold thrombosis |
| Type 5 | CABG-related MI | MI associated with coronary artery bypass grafting |
V. By Timing (Clinical)
- Acute MI - within 28 days of onset (first 7 days = "recent" MI)
- Old/healed MI - scar present, >28 days
Sequelae and Complications
The sequelae of MI are shown in the pathophysiology diagram:
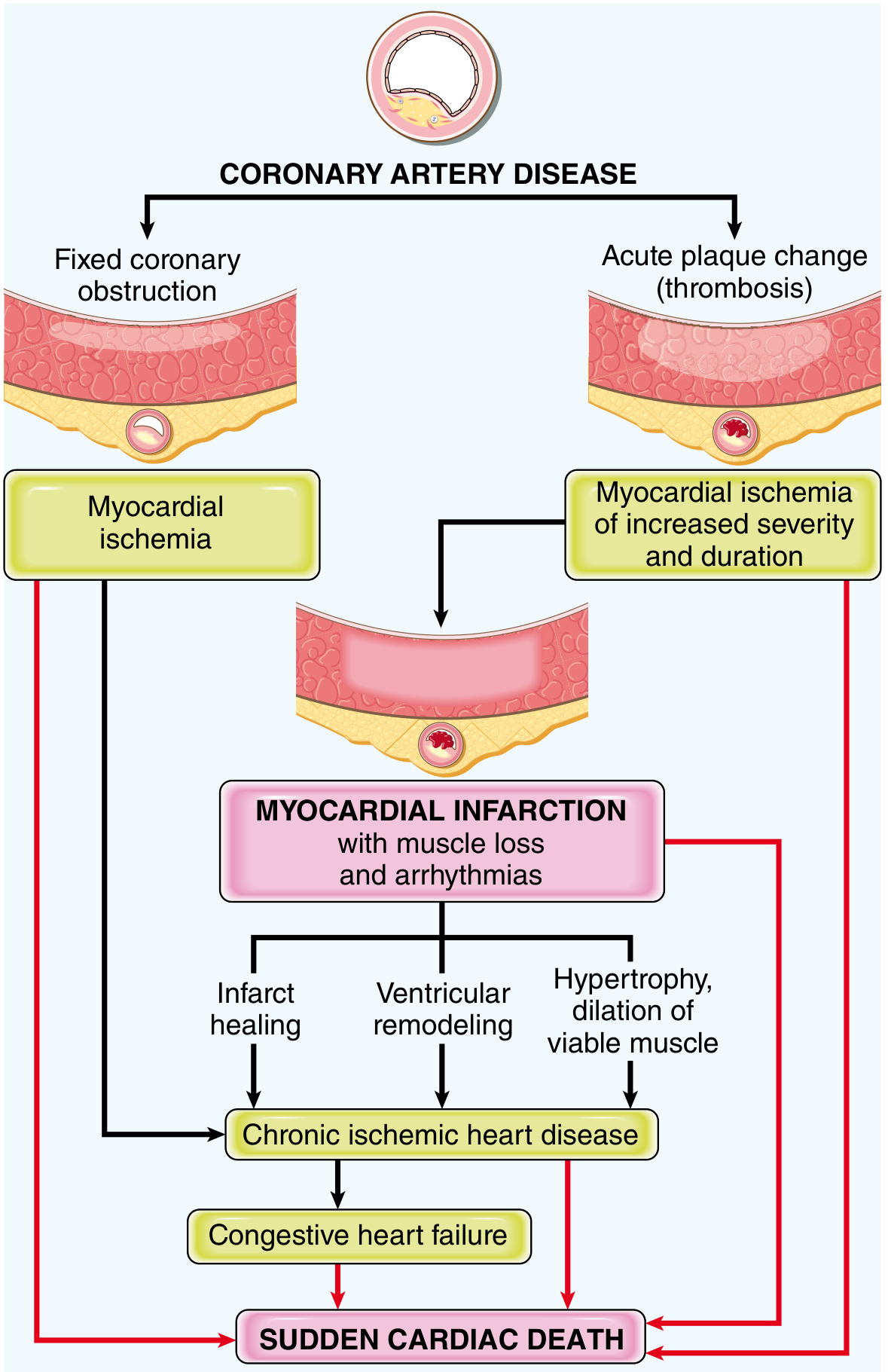
Major complications include:
- Ventricular rupture (3-7 days; free wall, VSD, papillary muscle)
- Papillary muscle dysfunction/rupture → acute mitral regurgitation
- Ventricular aneurysm - fibrous wall, paradoxical pulsation, mural thrombus
- Mural thrombus → systemic embolism
- Arrhythmias - most common cause of early death (VF)
- Pericarditis (Dressler syndrome - weeks later)
- Heart failure / cardiogenic shock
- Infarct extension or reinfarction
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (pp. 511-519); Robbins & Kumar Basic Pathology (p. 318); Sabiston Textbook of Surgery - Classification of AMI (Fourth Universal Definition)
Can you give me exam oriented concise answer
Myocardial Infarction — Exam-Oriented Concise Answer
Definition
Death of cardiac muscle due to prolonged ischemia (>20-40 min of severe ischemia).
Pathogenesis
The Core Sequence (90% of cases)
Atherosclerotic plaque
↓
Acute plaque disruption (rupture/erosion)
↓
Platelet adhesion → aggregation → activation
(releases TXA2, ADP, serotonin → vasospasm)
↓
Coagulation cascade activation (tissue factor)
↓
Complete coronary thrombosis
↓
Prolonged ischemia → NECROSIS
Key Points to Remember
- Culprit plaque often had <70% stenosis before rupture
- Thrombus visible in 90% if angiography done within 4 hrs
- ~10% of MIs occur without atherothrombosis (vasospasm, embolism, vasculitis, cocaine)
Myocardial Response to Ischemia
| Event | Time |
|---|---|
| Loss of contractility | < 2 min |
| Reversible injury | Up to 20-30 min |
| Irreversible necrosis begins | 20-40 min |
| Complete infarction | 6-12 hrs |
- Necrosis starts subendocardially (most vulnerable - last to receive blood, highest intramural pressure)
- Progresses outward = wavefront phenomenon
Reperfusion Injury (4 mechanisms - must know)
- Mitochondrial dysfunction → apoptosis
- Ca²⁺ overload → myocyte hypercontracture → contraction band necrosis
- Free radicals (O₂•⁻, H₂O₂, •OH) → membrane damage
- Neutrophil aggregation → "no-reflow" phenomenon
Classification
1. By Depth (Anatomical)
| Type | Features |
|---|---|
| Transmural | Full-thickness; permanent occlusion; → STEMI |
| Subendocardial | Inner 1/3 - 1/2 only; partial/transient occlusion; → NSTEMI |
| Microinfarction | Tiny foci; small vessel disease, vasospasm |
2. By ECG (Clinical)
| Type | ECG Change | Mechanism |
|---|---|---|
| STEMI | ST elevation → Q waves | Complete transmural occlusion |
| NSTEMI | ST depression / T changes; no Q waves | Partial/subendocardial; troponin ↑ |
| Unstable angina | Transient changes; no troponin rise | No necrosis |
3. By Coronary Territory
| Artery | Frequency | Area Infarcted |
|---|---|---|
| LAD | 40-50% | Anterior LV, anterior septum, apex |
| RCA | 30-40% | Inferior/posterior LV, posterior septum, RV |
| LCX | 15-20% | Lateral LV |
4. Universal Definition - Types 1-5 (High-yield)
| Type | Cause |
|---|---|
| Type 1 | Spontaneous - plaque rupture + thrombosis |
| Type 2 | Supply-demand mismatch (spasm, anemia, tachycardia, hypotension) |
| Type 3 | Sudden death before biomarkers obtained |
| Type 4a/b | PCI-related / Stent thrombosis |
| Type 5 | CABG-related |
Morphological Evolution (Exam favorite)
| Time | Gross | Microscopy |
|---|---|---|
| 0-4 hr | Nothing visible | Wavy fibers, coagulative necrosis beginning |
| 4-12 hr | Dark mottling | Early coagulative necrosis, edema |
| 12-24 hr | Dark mottling | Pyknotic nuclei, hypereosinophilia, neutrophils begin |
| 1-3 days | Yellow-tan center | Peak neutrophil infiltrate, loss of nuclei/striations |
| 3-7 days | Soft, yellow-tan; red border | Neutrophils dying; macrophages appear; granulation tissue begins |
| 7-10 days | Maximally soft | Granulation tissue prominent |
| 2-8 wks | Gray-white scar forming | Collagen deposition ↑ |
| >2 months | Dense white scar | Complete fibrosis |
Mnemomic for neutrophils → macrophages → granulation tissue → scar: "Never Mind Getting Sick" (Neutrophils → Macrophages → Granulation → Scar)
Complications (by timing)
| Timing | Complication |
|---|---|
| 0-24 hrs | Arrhythmias (VF) - most common cause of early death |
| 1-3 days | Cardiogenic shock |
| 3-7 days | Free wall rupture (→ tamponade), VSD, papillary muscle rupture (→ acute MR) |
| Weeks | Ventricular aneurysm, mural thrombus, Dressler syndrome (pericarditis) |
| Long-term | Chronic heart failure, sudden cardiac death |
High-yield one-liners:
- Earliest detectable necrosis marker in blood: Troponin (rises 3-4 hrs, peaks 24-48 hrs, persists 7-10 days)
- Earliest histologic change: Wavy fibers (0.5-4 hrs)
- Earliest gross change: Dark mottling (4-12 hrs; TTC stain can show pale unstained zone)
- Reperfusion hallmark: Contraction band necrosis
- "No-reflow" caused by: Neutrophil microvascular plugging
Chronic lymphocytic leukemia. Etiology. Pathogenesis. Laboratory diagnostics. Main clinical syndromes. Treatment principles. Prognosis. Concise exam oriented answer
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I have all the material needed. Here is the concise exam-oriented answer:
Chronic Lymphocytic Leukemia (CLL) — Exam-Oriented Concise Answer
Definition
Monoclonal proliferation of mature, functionally incompetent B lymphocytes (CD5+ B cells) in blood, bone marrow, lymph nodes, spleen, and liver. Defined by absolute lymphocyte count ≥5 × 10⁹/L of clonal B cells. CLL and Small Lymphocytic Lymphoma (SLL) are the same disease - different presentations.
Etiology & Risk Factors
| Factor | Detail |
|---|---|
| Age | Median diagnosis age 70; rare <40 yrs |
| Sex | Male:Female = 2:1 |
| Race | Most common in Caucasians; rare in Asians |
| Genetics | Most familial leukemia - 8.5x risk in first-degree relatives; >40 low-penetrance SNPs |
| Agent Orange | Officially recognized risk (Vietnam/Middle East veterans) |
| Radiation | NOT associated (unlike most other leukemias) |
| Infections | No established viral link |
| Precursor state | Monoclonal B-cell lymphocytosis (MBL) → ~1-2%/year progression to CLL |
High-yield: CLL is the most common leukemia in the Western world. It is NOT linked to radiation.
Pathogenesis
Cell of Origin
Mature B cell, immunophenotypically resembling a memory B cell (CD5+/CD23+).
Core Mechanisms
1. B-Cell Receptor (BCR) Signaling - Central driver
- CLL cells show tonic BCR activation → antiapoptotic signaling → tumor survival
- BCR drives NF-kB and PI3K/AKT pathways → proliferation + resistance to apoptosis
- BTK (Bruton's Tyrosine Kinase) is key downstream mediator → target of ibrutinib
2. IGHV Mutation Status - Key prognostic divider
| IGHV Status | Frequency | Course |
|---|---|---|
| Mutated (≥2% from germline) | ~60% | Indolent, good prognosis |
| Unmutated (<2% from germline) | ~40% | Aggressive, rapid progression, shorter survival |
3. Cytogenetic Abnormalities (detected by FISH)
| Abnormality | Frequency | Prognosis |
|---|---|---|
| del(13q14) | 50-55% | Favorable (disrupts miR-15a/16-1 → BCL2 overexpression) |
| Trisomy 12 | 20% | Intermediate |
| del(11q22) - ATM gene | 20% | Unfavorable - bulky nodes |
| del(17p13) - TP53 gene | 10% | Worst prognosis - therapy resistant |
4. Antiapoptosis via BCL-2
- miR-15a and miR-16-1 (deleted in del13q) normally suppress BCL-2
- Loss → BCL-2 overexpression → CLL cells accumulate (fail to die, not rapid proliferation)
- BCL-2 is the target of venetoclax
5. Key Somatic Mutations (adverse)
- TP53 mutation (independent of del17p) → chemorefractoriness
- NOTCH1 - associated with unmutated IGHV, Richter transformation
- SF3B1 - adverse; splicing factor
- ATM - DNA damage response defect
Laboratory Diagnostics
Peripheral Blood Smear (Pathognomonic finding)
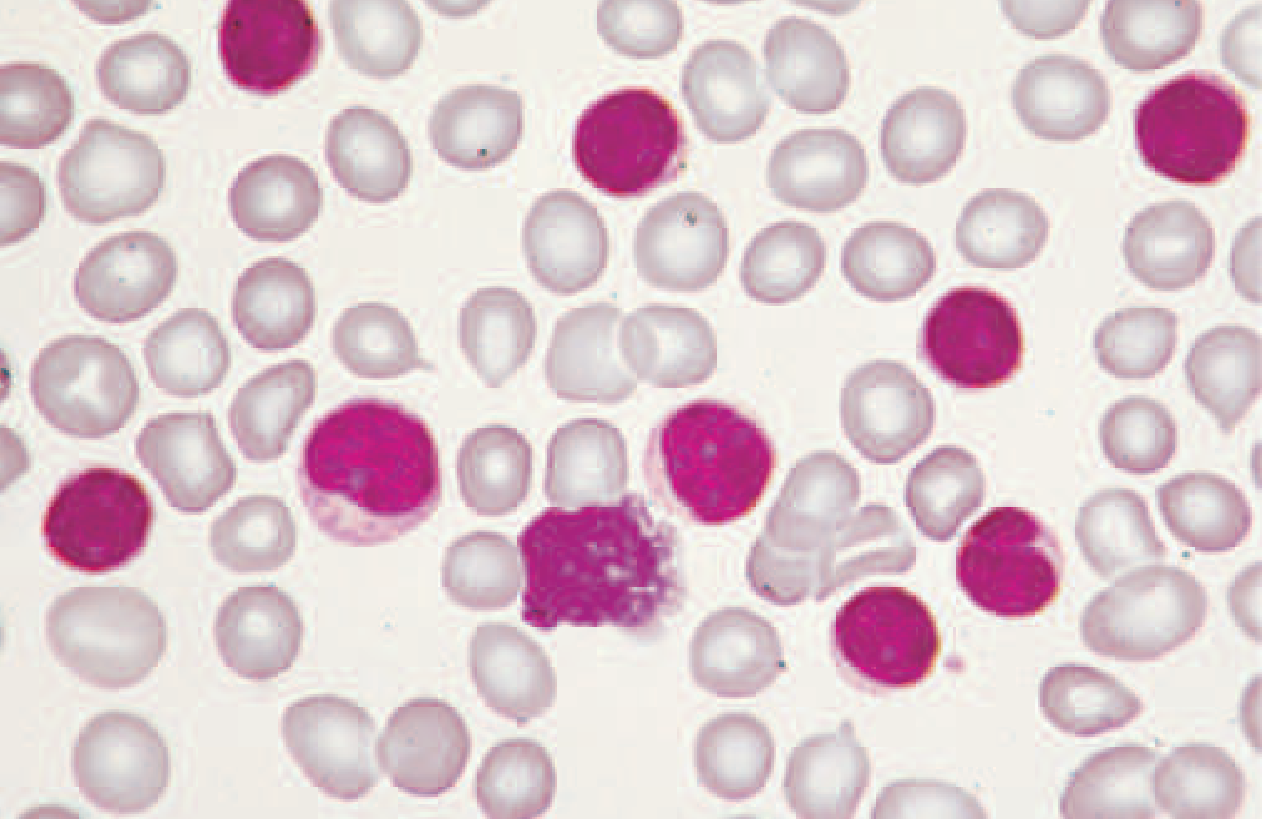
- Absolute lymphocytosis ≥5 × 10⁹/L
- Small, mature-appearing lymphocytes with clumped ("soccer ball") chromatin, scant cytoplasm
- Smudge/smear cells (Gumprecht shadows) - fragile CLL cells crushed during slide preparation - pathognomonic
- ± anemia, thrombocytopenia in advanced disease
Flow Cytometry (Immunophenotype) - ESSENTIAL for diagnosis
| Marker | CLL | Significance |
|---|---|---|
| CD5 | + | Aberrant (normally T-cell marker) |
| CD19, CD23 | + | B-cell lineage |
| CD20 | + (dim) | Dimly positive |
| Surface Ig | + (dim) | Single light chain (κ or λ) - monoclonal |
| CD10, Cyclin D1 | negative | Differentiates from follicular, mantle cell |
| LEF1 | + | Differentiates from other small B-cell lymphomas |
Mnemonic for CLL phenotype: "5, 19, 23, dim 20" = CD5+, CD19+, CD23+, CD20 dim
Bone Marrow Biopsy
- Diffuse/nodular/interstitial infiltration of small lymphocytes
- Pattern matters: diffuse = worse prognosis
Key Laboratory Tests
| Test | Purpose |
|---|---|
| FISH panel | del(13q), del(11q), del(17p), trisomy 12 - prognosis |
| IGHV mutational status | Key prognostic marker |
| TP53 mutation (NGS) | Guides therapy (BTK inhibitor vs chemo) |
| β2-microglobulin | Elevated = worse prognosis (tumor burden) |
| Serum immunoglobulins | Hypogammaglobulinemia (→ infection risk) |
| Stimulated karyotype | Complex karyotype = poor prognosis |
| Direct Coombs test | Screen for AIHA (autoimmune hemolytic anemia) |
Main Clinical Syndromes
1. Asymptomatic Lymphocytosis (most common presentation)
- ~70-80% diagnosed incidentally on routine CBC
- No symptoms, no treatment required
2. Lymphoproliferative Syndrome
- Lymphadenopathy - cervical, axillary, inguinal; non-tender, rubbery
- Splenomegaly - left upper quadrant fullness, early satiety
- Hepatomegaly (less common)
3. Bone Marrow Failure / Cytopenias
- Anemia - normocytic (marrow infiltration) OR hemolytic (AIHA - 10-25% of patients)
- Thrombocytopenia - marrow infiltration OR immune (ITP)
- Neutropenia - infection risk
4. Immunodeficiency Syndrome
- Hypogammaglobulinemia - recurrent bacterial infections (sinusitis, pneumonia, UTIs)
- Impaired T-cell function → viral and fungal infections
- Infections = leading cause of death (~30-50% of deaths)
5. Autoimmune Complications
- AIHA (autoimmune hemolytic anemia) - most common
- ITP (immune thrombocytopenic purpura)
- Pure red cell aplasia (rare)
6. Richter Transformation (~5-10%)
- Transformation to diffuse large B-cell lymphoma (most common) or Hodgkin lymphoma
- Presents: rapidly enlarging nodes, fever, weight loss, rising LDH
- Median survival after transformation: 3-6 months
Staging
Rai Staging System (USA)
| Stage | Features | Risk |
|---|---|---|
| 0 | Lymphocytosis only | Low |
| 1 | + Lymphadenopathy | Intermediate |
| 2 | + Splenomegaly/hepatomegaly | Intermediate |
| 3 | + Anemia (Hb <11 g/dL) | High |
| 4 | + Thrombocytopenia (<100,000/μL) | High |
Binet Staging System (Europe)
| Stage | Features | Median Survival |
|---|---|---|
| A | <3 lymphoid areas involved; no anemia/thrombocytopenia | >10 years |
| B | ≥3 lymphoid areas; no anemia/thrombocytopenia | 5-7 years |
| C | Anemia (Hb <10) and/or thrombocytopenia (<100k) | 2-3 years |
Treatment Principles
When to Treat (Indications - NOT stage alone)
- Hb <10 g/dL or platelets <100,000/μL (marrow failure)
- Symptomatic/bulky lymphadenopathy (>10 cm) or splenomegaly
- Constitutional symptoms (B symptoms: fever, night sweats, weight loss)
- Lymphocyte doubling time <6 months
- Difficult-to-control AIHA or ITP
Important: Lymphocytosis alone is NOT an indication to treat.
First-Line Treatment (Current Standard - targeted therapy era)
For most patients (del17p/TP53 wild-type or mutant):
| Agent | Mechanism | Notes |
|---|---|---|
| Ibrutinib (BTK inhibitor) | Blocks BTK → BCR signaling → apoptosis | First-line; oral; continuous |
| Acalabrutinib | 2nd-gen BTK inhibitor | Better tolerability than ibrutinib |
| Venetoclax + obinutuzumab | BCL-2 inhibitor + anti-CD20 | Fixed duration (12 months); preferred for fit patients |
| Zanubrutinib | 3rd-gen BTK inhibitor | Fewer off-target effects |
For del(17p) / TP53 mutation (chemo-resistant):
- BTK inhibitors (ibrutinib, acalabrutinib, zanubrutinib) - preferred
- Venetoclax combinations
- Avoid chemoimmunotherapy (FCR) - ineffective
Older chemoimmunotherapy (now 2nd/3rd line for fit, mutated IGHV, low-risk):
- FCR: Fludarabine + Cyclophosphamide + Rituximab
- BR: Bendamustine + Rituximab
Supportive Treatment
- IVIg - for recurrent infections with hypogammaglobulinemia
- Vaccinations - influenza, COVID-19, pneumococcal, zoster (recombinant; avoid live)
- Steroids - for AIHA/ITP
- Allogeneic SCT - considered in young patients with high-risk disease
Prognosis
| Factor | Favorable | Unfavorable |
|---|---|---|
| IGHV | Mutated (≥2%) | Unmutated (<2%) |
| FISH | del(13q) alone | del(17p), del(11q) |
| TP53 | Wild-type | Mutated |
| Karyotype | Normal / simple | Complex (≥3 abnormalities) |
| ZAP-70 | Negative | Positive |
| CD38 | <30% | ≥30% |
| β2-microglobulin | Normal | Elevated |
| Lymphocyte doubling time | >12 months | <6 months |
| Stage | Rai 0-1 / Binet A | Rai 3-4 / Binet C |
Overall survival: 5-year OS ~92% (2015 data); markedly improved with targeted therapies. ~30% of patients never need treatment.
Leading causes of death: Infections (~30-50%), Richter transformation, second malignancies, progressive CLL.
High-Yield One-Liners
- CLL = most common leukemia in the Western world
- NOT linked to radiation (unique among leukemias)
- Smudge cells on PBS = pathognomonic
- Immunophenotype: CD5+, CD19+, CD23+, CD20 dim, surface Ig dim
- del(13q) = best prognosis; del(17p)/TP53 = worst prognosis
- BCL-2 overexpression → cells accumulate (don't proliferate rapidly)
- Treatment target: BTK (ibrutinib) and BCL-2 (venetoclax)
- Richter transformation = CLL → DLBCL; very poor prognosis
- AIHA = most common autoimmune complication
Sources: Harrison's Principles of Internal Medicine 22E (2025); Goldman-Cecil Medicine; Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.