Interpretation
HPLC HbA2/F method HbF 36% elevated interpretation newborn hemoglobin
HPLC Hemoglobin Fractionation Report — Interpretation
Peak Results
| Peak | Retention Time | Area % |
|---|---|---|
| A1a | 0.20 min | 41.9% |
| F (HbF) | 0.37 min | 36.0% ← flagged (*) |
| A0 (HbA) | 1.78 min | 26.0% |
| HbA2 | Not detected / not reported | — |
Key Finding: Markedly Elevated HbF at 36%
- Newborn: 50–80%
- 6 months: ~8%
- 1 year: <5%
- Adults: <2%
Differential Diagnosis for HbF 36%
What's Missing / Noteworthy
- HbA2 is not reported — on the D-10 HbA2/F program, HbA2 is typically quantified separately. Its absence from the peak table is notable; it may be subsumed into another peak or below detection. A normal HbA2 (<3.5%) + elevated HbF would favor HPFH or δβ-thalassemia over β-thalassemia trait (which raises HbA2 to 3.5–7%).
- The A1a fraction at 41.9% is very high — in adults this is abnormal (normal ~1.5–2.5%). In a newborn, early-eluting peaks behave differently and the A1a window may capture non-specific or modified Hb fractions. This warrants correlation with the patient's age.
- The total area is 23,324 with asterisk (*), suggesting the instrument flagged this run as potentially unreliable or outside calibration range.
Bottom Line
| If patient is a neonate | Normal transitional pattern, but A1a elevation and low-for-age HbF warrant repeat at 6–12 months |
|---|---|
| If patient is a child/adult | HbF of 36% is markedly abnormal; most likely HPFH (heterozygous) or δβ-thalassemia; obtain CBC, reticulocyte count, serum ferritin, and repeat HPLC with full hemoglobin panel including HbA2 quantitation |
2 year baby girl
HbF 36% HPLC 2 year old child beta thalassemia HPFH differential diagnosis
homozygous delta beta thalassemia HbF 36% child HPFH HPLC
Revised Interpretation — 2-Year-Old Girl
The Critical Abnormality
HPLC Pattern Analysis
| Fraction | Result | Expected at age 2 | Interpretation |
|---|---|---|---|
| HbF | 36% | <2% | Severely elevated |
| HbA (A0) | 26% | >97% | Markedly reduced |
| HbA2 | Not reported | 2–3.5% | Absent/not quantified |
| HbA1a | 41.9% | ~1.5% | Abnormally elevated |
Most Likely Diagnosis: β⁺-Thalassemia (Intermedia)
- HbF 20–40% + HbA present (26%) + reduced HbA2 or normal HbA2
"In Black people with β⁺-thalassemia, clinical features are less severe (thalassemia intermedia), and transfusion is usually unnecessary; HbF is 20% to 40%, HbA2 is 2%–5%, and the rest is HbA." — Henry's Clinical Diagnosis, 23rd ed.
Differential Diagnosis
| Condition | HbF | HbA | HbA2 | Likelihood |
|---|---|---|---|---|
| β⁺-Thalassemia intermedia | 20–40% | Present | 2–5% | ✅ Most likely |
| β⁰-Thalassemia major | 60–98% | Absent | ~2% | ❌ Less likely (HbA is present) |
| δβ-Thalassemia (heterozygous) | 5–20% | Present | Normal | ❌ HbF too high |
| HPFH (heterozygous) | 15–35% | Present | Normal | ⚠️ Possible — clinically benign |
| HPFH (homozygous) | ~100% | Absent | — | ❌ HbA is present |
What You Must Do Next
- CBC with indices — expect microcytic hypochromic anemia (low MCV, MCH), low Hb
- Serum ferritin + iron studies — to exclude concurrent iron deficiency (which can falsely lower HbA2 and alter the HPLC pattern)
- Repeat HPLC with HbA2 quantitation — the D-10 HbA2/F program should give HbA2; if missing, rerun or request a dedicated HbA2 program
- Parental HPLC — essential to establish inheritance pattern (both parents likely carriers)
- Molecular/DNA testing — confirm the specific β-globin mutation (β⁺ vs β⁰) and determine if compound heterozygosity is present
- Clinical assessment — check growth, spleen size, Hb level; if Hb <7 g/dL with symptoms, may need transfusion support despite "intermedia" label
Bottom Line

CBC o this patient
CBC Interpretation — 2-Year-Old Girl
Values at a Glance
| Parameter | Result | Reference (2yr girl) | Status |
|---|---|---|---|
| WBC | 11.0 × 10³/µL | 6–17 | ✅ Normal |
| Lymph# | 7.4 × 10³/µL | 3–9.5 | ✅ Normal |
| Lymph% | 67.1% | 45–75% | ✅ Normal |
| Gran% | 28.0% | — | ⬇️ Low (relative) |
| RBC | 4.42 × 10⁶/µL | 3.7–5.3 | ✅ Normal |
| HGB | 7.7 g/dL | 11.5–13.5 | 🔴 Severely LOW |
| HCT | 26.2% | 34–40% | 🔴 Severely LOW |
| MCV | 59.4 fL | 72–86 | 🔴 Severely LOW (microcytic) |
| MCH | 17.5 pg | 24–30 | 🔴 Severely LOW (hypochromic) |
| MCHC | 29.4 g/dL | 32–36 | 🔴 LOW |
| RDW-CV | 16.6% | 11.5–14.5 | 🔴 HIGH (anisocytosis) |
| RDW-SD | 34.6 fL | 35–56 | ⬇️ Low-normal |
| PLT | 344 × 10³/µL | 150–400 | ✅ Normal |
| PDW | 15.0 | <17 | ✅ Borderline |
Key Findings
1. Severe Microcytic Hypochromic Anemia
- Hb 7.7 g/dL — significantly below the 11.5 g/dL lower limit for age. This is moderate-to-severe anemia.
- MCV 59.4 fL — deeply microcytic (normal for age ~72–86 fL). A drop this profound rules out pure iron deficiency as the sole cause; MCV this low points strongly to thalassemia.
- MCH 17.5 pg — severely hypochromic, consistent with deficient hemoglobin synthesis.
- MCHC 29.4 g/dL — reduced, confirming hypochromia.
2. Elevated RDW-CV (16.6%)
- Indicates significant anisocytosis (variation in RBC size).
- In thalassemia, RDW is typically mildly-to-moderately elevated (unlike iron deficiency, which can raise it more markedly). A value of 16.6% fits well.
- The RDW-SD is paradoxically low-normal (34.6 fL) because the RBC population is uniformly shifted to a very small size — the curve is narrow but left-shifted, which the histogram confirms (tight peak around 60–80 fL).
3. RBC Histogram
4. WBC and Platelet: Normal
- WBC 11.0 — normal for age; mild lymphocyte predominance is physiological at 2 years.
- PLT 344 — normal; reactive thrombocytosis can occur in thalassemia with hemolysis (not prominent here).
Integrated Diagnosis
| Feature | This Patient | β-Thalassemia Intermedia |
|---|---|---|
| Hb | 7.7 g/dL | 7–10 g/dL |
| MCV | 59.4 fL | 55–70 fL |
| MCH | 17.5 pg | <27 pg |
| HbF | 36% | 20–40% |
| HbA | 26% | Present |
| HbA2 | Not quantified | 2–5% |
Urgent Next Steps
- Check if this Hb is baseline or post-illness — acute infection can drop Hb further
- Serum ferritin + iron/TIBC — concurrent iron deficiency is common in thalassemia and worsens anemia
- Reticulocyte count — expect elevated (compensatory erythropoiesis)
- Peripheral blood smear — expect target cells, tear drops, hypochromic microcytes, nucleated RBCs
- HbA2 quantitation (separate program or repeat HPLC)
- Parental CBC + HPLC — both parents should be tested
- Hematology referral — to decide on transfusion threshold and long-term management (regular transfusion ± chelation vs. hydroxyurea vs. curative stem cell transplant)
If Hb <7 g/dL or symptomatic (pallor, failure to thrive, tachycardia), transfusion should not be delayed.

Correct it
- Iron profile (serum ferritin, serum iron, TIBC) — to exclude concurrent iron deficiency
- HbA2 quantitation (repeat HPLC or dedicated HbA2 program)
- Reticulocyte count
- Parental CBC and HPLC (both parents) — for thalassemia trait confirmation
- Haematology referral for transfusion assessment and long-term management
Corrections Made
| # | Original Error | Correction |
|---|---|---|
| 1 | "mild anisopoikilocytosis" | Should be moderate — the degree of microcytosis (MCV 59.4) and hypochromia (MCH 17.5) in thalassemia produces more than mild change |
| 2 | Spelling: "thalaseemia intermedia" | Beta-thalassemia intermedia (double 's' not needed; single 's') |
| 3 | Spelling: "ramge" in platelet section | range |
| 4 | Advice to perform HPLC | HPLC has already been done — advising it again is an error. Replace with HbA2 quantitation + parental studies |
| 5 | Impression lacks Hb level context | Should note Hb 7.7 g/dL as moderate-to-severe anaemia |
| 6 | No mention of nucleated RBCs | Expected finding in thalassemia intermedia — should be noted if seen on smear |
| 7 | Impression says "probable" without specifics | Should reference the actual HPLC values (HbF 36%) to justify the diagnosis |
Reticulocyte count is 1.1%
Reticulocyte Count Interpretation
Is 1.1% Normal or Abnormal Here?
Step 1 — Corrected Reticulocyte Count
Step 2 — Reticulocyte Production Index (RPI)
Interpretation
| Index | Value | Threshold | Meaning |
|---|---|---|---|
| Raw retic% | 1.1% | >2% expected | Inadequate |
| CRC | 0.8% | >2% = adequate | Inadequate |
| RPI | 0.53 | >2 = adequate response | Severely inadequate |
What This Means Clinically
Defective β-globin synthesis → excess α-chains precipitate → RBC precursors destroyed in the marrow before reaching circulation → low retic count despite high erythroid activity in the bone marrow
Contrast with Iron Deficiency
| Feature | This Patient | Pure Iron Deficiency |
|---|---|---|
| Retic count | Low (0.53 RPI) | Low initially, rises with treatment |
| Mechanism | Ineffective erythropoiesis | Insufficient substrate |
| Marrow | Hyperplastic | Hyperplastic but with low iron stores |
| Response to iron | No improvement in Hb | Hb rises 1–2 g/dL per week |
Bottom Line
sebaceous cyst vs epidermal inclusion cyst ??
Sebaceous Cyst vs. Epidermal Inclusion Cyst
The Core Issue
"The term 'sebaceous cyst' is a misnomer and should be avoided because these cysts do not involve sebaceous glands, nor do they contain sebum." — Fitzpatrick's Dermatology, 9th ed.
"The only true sebaceous cyst is the steatocystoma. The term 'sebaceous cyst' is best avoided." — Dermatology 2-Volume Set, 5th ed. (Bolognia)
Side-by-Side Comparison
| Feature | Epidermal Inclusion Cyst (Epidermoid Cyst) | True "Sebaceous Cyst" (Steatocystoma) |
|---|---|---|
| Correct name | Epidermoid cyst / Epidermal inclusion cyst | Steatocystoma multiplex/simplex |
| Origin | Plugged pilosebaceous infundibulum (upper follicle); or traumatic implantation of epidermis | Sebaceous duct / sebaceous gland |
| Contents | Keratin — white/yellow cheesy, foul-smelling debris | Sebum — oily/liquid yellowish material |
| Wall lining (histology) | Stratified squamous epithelium with granular layer | Thin epithelium without granular layer; sebaceous lobules in wall |
| Punctum | Present (central dark punctum — plugged follicle) | Absent |
| Location | Face, neck, upper chest, upper back, scalp | Chest, axilla, scrotum (steatocystoma multiplex is diffuse) |
| Genetics | May occur in Gardner syndrome, Gorlin syndrome | Steatocystoma multiplex → KRT17 mutation (pachyonychia congenita type 2) |
| Smell on expression | Foul-smelling cheesy material | Odourless oily fluid |
| Common? | Very common | Uncommon |
| Malignant potential | Rare (BCC, SCC reported) | Very rare |
| Treatment | Complete excision of cyst wall | Excision; multiple lesions may be managed with CO₂ laser |
Histological Key Difference
| Epidermoid Cyst | Steatocystoma | |
|---|---|---|
| Granular layer | Present | Absent |
| Wall | Squamous epithelium (like skin surface) | Thin, corrugated epithelium with sebaceous gland lobules embedded in wall |
| Contents | Laminated orthokeratin | Amorphous sebaceous material |
Pilar Cyst (Trichilemmal Cyst) — Often Confused Too
| Feature | Epidermoid Cyst | Pilar Cyst |
|---|---|---|
| Origin | Follicular infundibulum | Hair follicle isthmus (trichilemma) |
| Granular layer | Present | Absent |
| Contents | Soft cheesy keratin | Hard, compact keratin (may calcify) |
| Location | Face, chest, back | Scalp (90%) |
| Punctum | Usually present | Usually absent |
| Genetics | Gardner syndrome | Autosomal dominant; PLCD1 gene |
Practical Summary
- In clinical practice, when you see a subcutaneous mobile nodule with a central punctum expressing cheesy material → call it an epidermal inclusion cyst (not sebaceous cyst).
- Reserve "sebaceous cyst" only for steatocystoma, which expresses oily fluid and has no punctum.
- On the histopathology report, the presence or absence of a granular layer is the key to distinguishing all three. — Fitzpatrick's Dermatology, 9th ed., p. 1842–1843

Check grammar and format
- Endometrium — Proliferative phase.
- Myometrium — Intramural leiomyoma and subserosal leiomyoma.
- Cervix — Chronic cervicitis.
Corrections Made
| # | Original | Corrected | Reason |
|---|---|---|---|
| 1 | "externally the uterus..." (lowercase) | "Externally, the uterus..." | Sentence must start with capital; comma added after introductory word |
| 2 | "A large subserosal fibroids is identified" | "A large subserosal fibroid is identified" | Subject-verb agreement: singular "fibroid" |
| 3 | "largest measuring 7.2x6.8x6.5" | "7.2 × 6.8 × 6.5 cm" | Use × symbol (not x) for dimensions; unit placed at end |
| 4 | "endo and ectocervix" | "endo**-** and ectocervix" | Correct use of suspended hyphen |
| 5 | "Section from Endomyometrium" | "Sections from the endomyometrium" | Lowercase (not a proper noun); add "the"; plural "Sections" consistent with rest |
| 6 | "hyperplasia/ atypia/ malignancy" | "hyperplasia, atypia, or malignancy" | Use commas and "or" in a list, not slashes |
| 7 | "Myometrium small leiomyoma." | "Myometrium shows a small leiomyoma." | Incomplete sentence — verb missing |
| 8 | "endocervical ling" | "endocervical lining" | Typographical error |
| 9 | "Intra Mural" | "Intramural" | One word, no space |
| 10 | "Sub Serosal" | "Subserosal" | One word; lowercase |
| 11 | Impression format: dashes with inconsistent spacing | Bullet-point list with consistent formatting | Standardised layout |
| 12 | "c/o Fibroid" | "C/o fibroid" | Lowercase "fibroid" (not a proper noun) |
| 13 | Dimension "12x10.9x7.8" | "12 × 10.9 × 7.8 cm" | Proper multiplication symbol with spaces |
Create a summary document of this histopathology report for the patient's file
APL H 06 26 Patient Summary
Word Document · DOCX
- Header — "Histopathology Report — Patient Summary" with subtitle marking it as confidential
- Report Details section — reference number, specimen type, indication, date
- Summary Table — colour-coded two-column table covering all key findings at a glance, with "NOT detected" for malignancy highlighted in red
- Gross Examination — corrected and complete paragraph
- Microscopic Examination — broken into three underlined subsections (Endometrium, Myometrium, Cervix)
- Final Impression / Diagnosis — bulleted diagnosis list with a prominent red "No evidence of malignancy" statement
- Signature block — for the reporting pathologist with date line
Endometrial hyperplasia grading and microscopy
endometrial hyperplasia microscopy histology simple complex atypical
Endometrial Hyperplasia — Classification, Grading & Microscopy
Classification Systems
1. WHO / Traditional System (4-tier)
| Type | Architecture | Cytologic Atypia | Risk of Progression to Carcinoma |
|---|---|---|---|
| Simple (cystic without atypia) | Dilated, cystic glands; increased gland:stroma ratio | None | 1% |
| Complex (adenomatous without atypia) | Crowded, budding, back-to-back glands | None | 3% |
| Atypical Simple | Dilated/simple glands | Present | 8% |
| Atypical Complex | Crowded, complex glands | Present | 29% |
2. WHO 2014 / EIN System (2-tier — currently recommended)
| Category | Equivalent (old) | Risk | Management |
|---|---|---|---|
| Hyperplasia without atypia | Simple + Complex (no atypia) | 1–3% | Progestins / surveillance |
| Atypical Hyperplasia / EIN (Endometrioid Intraepithelial Neoplasia) | Atypical simple + Atypical complex | 25–43% concurrent carcinoma at hysterectomy | Hysterectomy |
Microscopic Features
Hyperplasia WITHOUT Atypia (NEH)
- Increased gland-to-stroma ratio (crowded glands)
- Glands vary in size and shape — may be cystic/dilated
- Some intervening stroma retained (not purely back-to-back)
- Tubular or irregular gland outlines
- Epithelium is simple columnar with pseudostratification
- Nuclei are elongated, uniform, maintain polarity (perpendicular to basement membrane)
- No nuclear enlargement, no prominent nucleoli
- Mitoses may be increased but no atypia
- Evenly dispersed chromatin
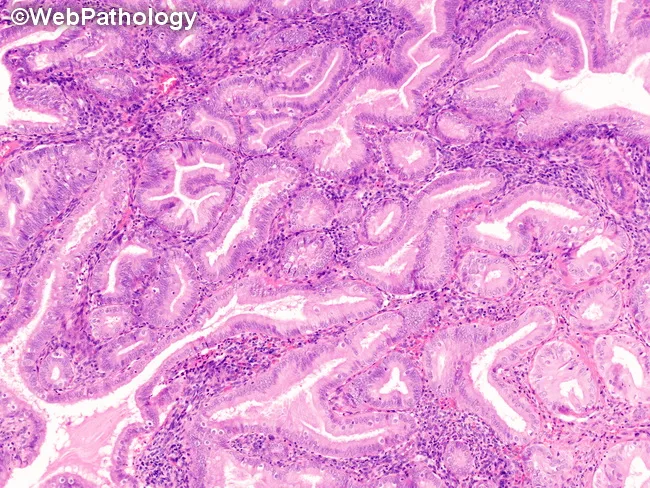
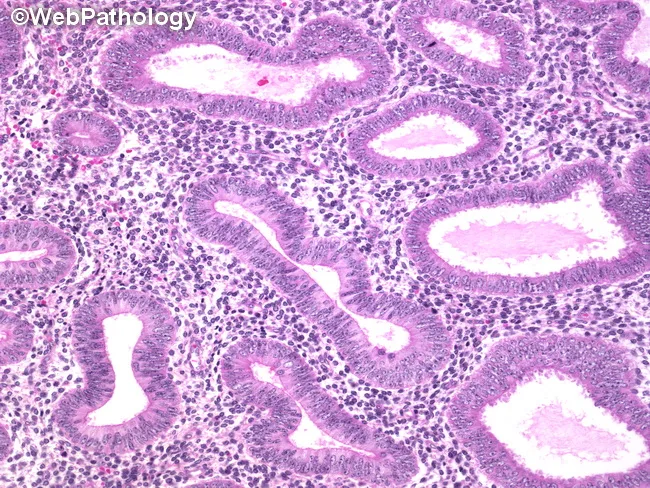
Atypical Hyperplasia / EIN
- Back-to-back glands with gland:stroma ratio >1
- Complex outlines — budding, branching, infolding
- Clonal outgrowth (focus >1 mm)
- Less intervening stroma
- Nuclear enlargement — large, round (lose elongated shape)
- Loss of nuclear polarity (no longer perpendicular to basement membrane)
- Vesicular (open) chromatin with irregular clumping
- Prominent nucleoli
- Increased nuclear-to-cytoplasmic ratio
- Cellular stratification
- High mitotic rate
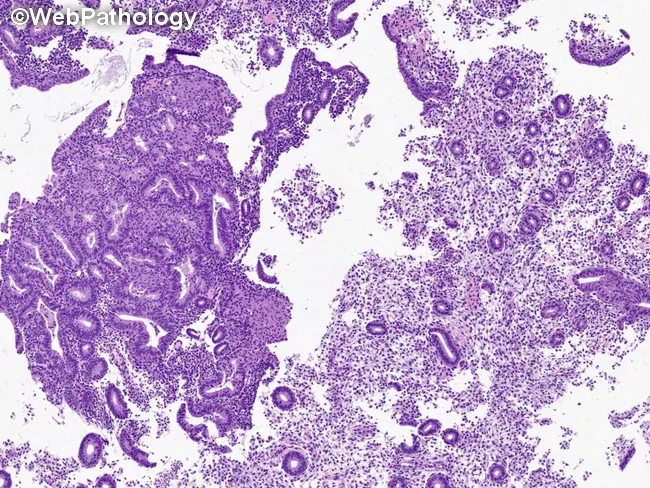
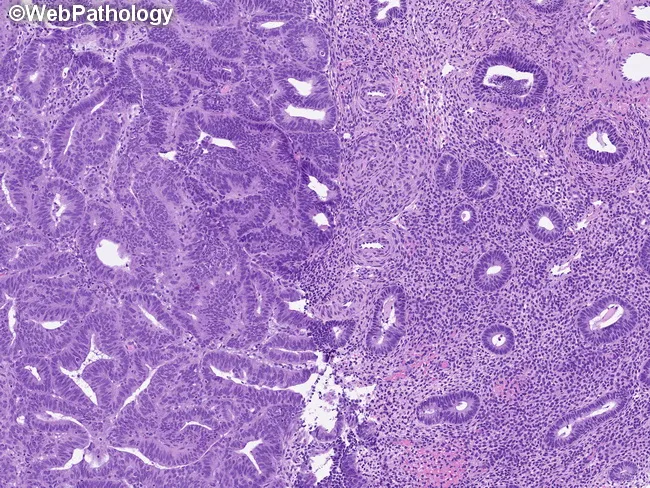
Key Distinguishing Feature: Atypia vs. No Atypia
| Feature | No Atypia | Atypia (EIN) |
|---|---|---|
| Nuclear shape | Elongated | Rounded/enlarged |
| Nuclear polarity | Preserved | Lost |
| Nucleoli | Inconspicuous | Prominent |
| Chromatin | Uniform | Vesicular, clumped |
| Gland:stroma | Increased | >1, back-to-back |
| Risk | 1–3% | 25–43% concurrent Ca |
Molecular Basis
- PTEN mutation — most common; found in >20% of hyperplasias (both with and without atypia) and 30–80% of endometrial carcinomas → early event in endometrial tumorigenesis
- PI3K/AKT pathway — overactivated when PTEN is lost; also amplifies estrogen receptor signalling
- Mismatch repair gene mutations — associated with Lynch syndrome and endometrioid carcinoma
- Cowden syndrome (germline PTEN mutation) — markedly elevated risk of endometrial carcinoma
Risk Factors (Unopposed Estrogen)
- Obesity (peripheral androgen → estrogen conversion)
- Anovulation / PCOS
- Perimenopausal anovulatory cycles
- Exogenous estrogen without progestin
- Granulosa cell tumours of the ovary
- Tamoxifen use
Clinical Importance
"Up to 40% of women with a diagnosis of atypical hyperplasia are found to have carcinoma when hysterectomy is performed." — Robbins & Kumar Pathologic Basis of Disease, 10th ed.
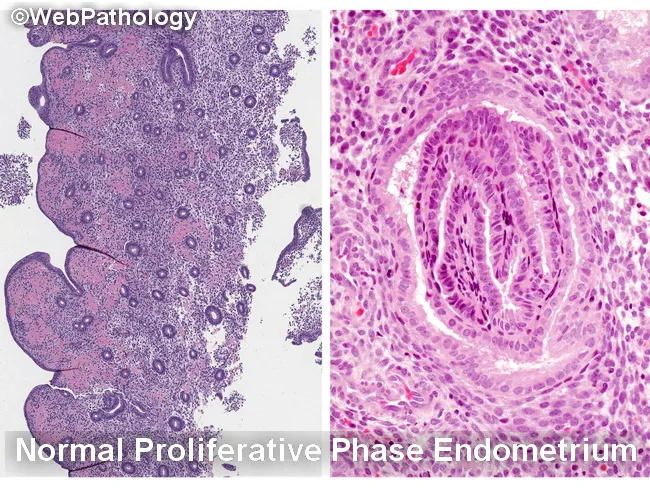
Is description of proliferative phase is correct in the image it uplodaded
proliferative phase endometrium histology tubular glands pseudostratification compact stroma
Report Description (from the image)
"Section from Endomyometrium reveals round tubular glands lined by cuboidal to columnar epithelium with pseudostratification at places. Stroma is compact and at places loose. There is no evidence of hyperplasia/atypia/malignancy." Impression: Proliferative Phase
What Is Correct ✅
| Feature Mentioned | Accuracy |
|---|---|
| Round tubular glands | ✅ Correct — proliferative phase glands are tubular/straight |
| Cuboidal to columnar epithelium | ✅ Correct |
| Pseudostratification | ✅ Correct — characteristic of proliferative phase |
| Stroma compact and at places loose | ✅ Acceptable — proliferative stroma is typically cellular and compact |
| No hyperplasia/atypia/malignancy | ✅ Correct statement to include |
What Is Missing / Should Be Added ❌
| Missing Feature | Why It Matters |
|---|---|
| Mitotic figures | The single most important hallmark of proliferative phase — active cell division driven by estrogen. Their presence confirms the phase. Must be mentioned |
| Nuclei elongated/pencil-shaped, basally oriented | Key cytologic feature of proliferative glands — nuclei are elongated and parallel to each other |
| No subnuclear vacuoles | Should be explicitly stated to exclude early secretory phase (Day 17) |
| Straight/mildly tortuous glands | Early proliferative = straight; late proliferative = slightly tortuous/wavy. Specifying helps date the phase |
| Gland:stroma ratio normal | Should state that there is no increased gland:stroma ratio (to exclude hyperplasia) |
| Stroma: spindle-shaped cells with scant cytoplasm | A descriptor of normal proliferative stroma |
Corrected / Complete Microscopic Description
"Sections from the endomyometrium reveal straight to mildly tortuous tubular glands lined by columnar epithelium with pseudostratification. The nuclei are elongated, basally oriented, with evenly dispersed chromatin. Mitotic figures are seen in the glandular epithelium. Subnuclear vacuolation is absent. The stroma is cellular, composed of spindle-shaped cells with scant cytoplasm, and is compact. The gland-to-stroma ratio is within normal limits. There is no evidence of hyperplasia, atypia, or malignancy."
What Proliferative Phase Looks Like