Write assignment on aldosterone
Reading File
Finding Sources
Finding Sources
Searching PubMed
aldosterone physiology regulation cardiovascular
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Aldosterone: A Comprehensive Assignment
1. Introduction
Aldosterone is the principal mineralocorticoid hormone in humans. It is a steroid hormone produced by the adrenal cortex, and its core physiological role is the maintenance of sodium and potassium balance, blood pressure, and extracellular fluid volume. It does this by acting on epithelial cells of the kidney, colon, salivary glands, and sweat glands. Beyond these classical roles, accumulating evidence shows that excess aldosterone exerts harmful non-epithelial effects on the cardiovascular system, kidneys, and central nervous system - a recognition that has shifted how clinicians think about mineralocorticoid receptor (MR) blockade.
2. Chemistry and Structure
Aldosterone is a C21 steroid derived from cholesterol. Its chemical name is 11β,21-dihydroxy-3,20-dioxo-4-pregnen-18-al. The defining structural feature is a characteristic C18 aldehyde group, from which the hormone takes its name. Unlike cortisol (which has a hydroxyl group at C17), aldosterone lacks a 17α-hydroxyl group, which is key to its selective mineralocorticoid rather than glucocorticoid activity.
- Molecular formula: C₂₁H₂₈O₅
- Molecular weight: 360.44 g/mol
- Plasma half-life: approximately 20 minutes
- Plasma levels (normal): approximately 0.006 μg/dL (0.17 nmol/L)
- Daily secretion rate: 50-200 μg/day
It is only weakly bound to plasma proteins (primarily albumin), in contrast to cortisol, which is tightly bound to cortisol-binding globulin. This short half-life means its secretory rate directly determines its biological effect.
(Ganong's Review of Medical Physiology, 26th Ed.)
3. Biosynthesis
3.1 Site of Synthesis
Aldosterone is synthesized exclusively in the zona glomerulosa - the outermost layer of the adrenal cortex. This anatomical specificity is enforced at the enzymatic level. The zona glomerulosa uniquely expresses aldosterone synthase (CYP11B2) but lacks the enzyme 17α-hydroxylase (expressed only in the zona fasciculata). This means the zona glomerulosa cannot produce cortisol or androgens, only aldosterone.
3.2 Biosynthetic Pathway
The pathway proceeds from cholesterol and is illustrated in the diagram below:
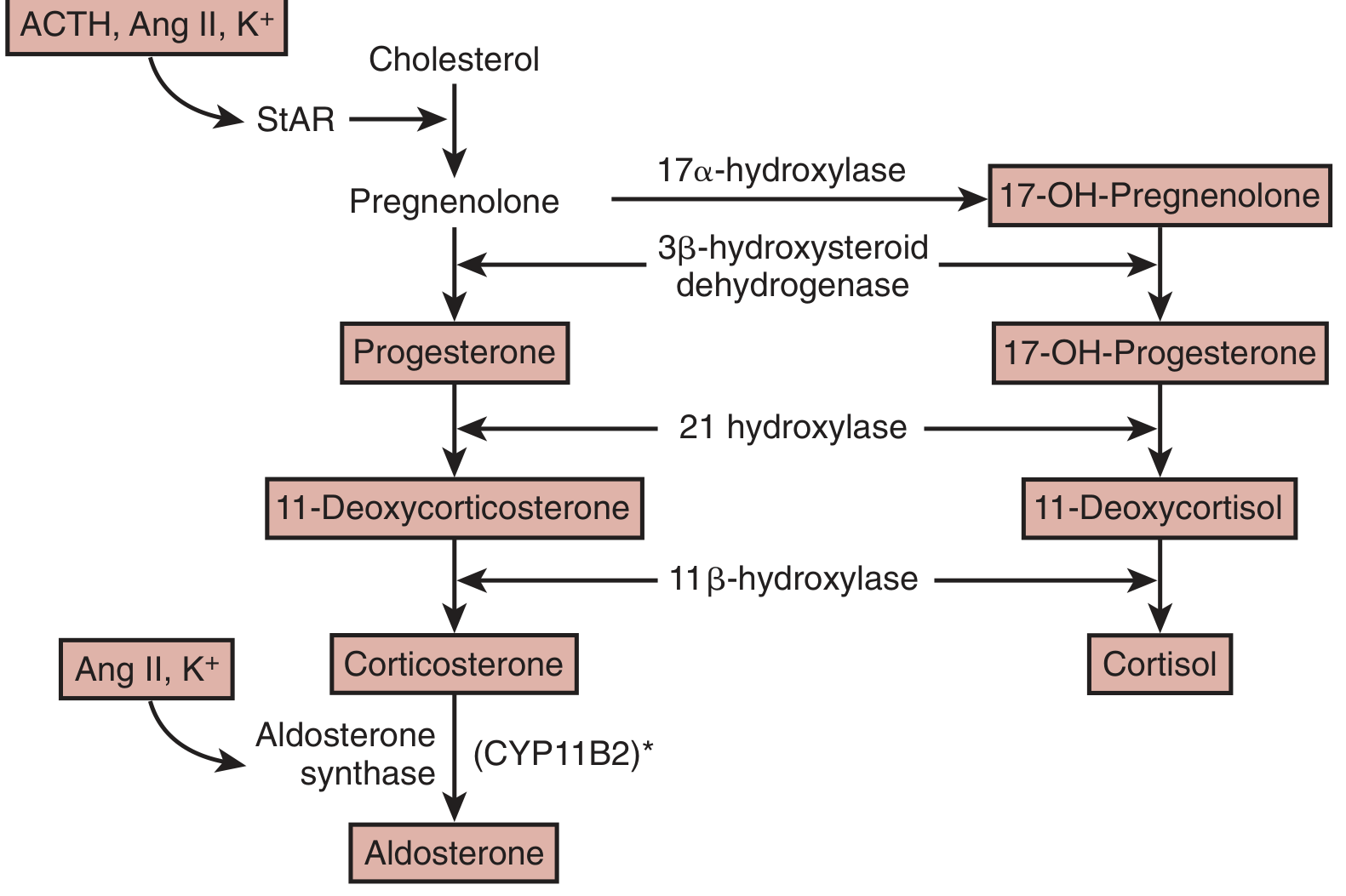
Steps:
- Cholesterol → Pregnenolone - catalyzed by side-chain cleavage enzyme (CYP11A1); requires transport of cholesterol into the inner mitochondrial membrane by StAR (steroidogenic acute regulatory protein)
- Pregnenolone → Progesterone - by 3β-hydroxysteroid dehydrogenase (3β-HSD)
- Progesterone → 11-Deoxycorticosterone (DOC) - by 21-hydroxylase (CYP21)
- DOC → Corticosterone - by 11β-hydroxylase (CYP11B1, in the fasciculata) / aldosterone synthase (in the glomerulosa)
- Corticosterone → Aldosterone - by aldosterone synthase (CYP11B2) in a unique three-step reaction:
- 11β-hydroxylation
- 18-hydroxylation
- 18-methyl oxidation → yielding the C18-aldehyde characteristic of aldosterone
This final three-step conversion is performed exclusively by CYP11B2, which is absent from the zona fasciculata, explaining why cortisol synthesis cannot generate aldosterone.
(Brenner and Rector's The Kidney, 2-Volume Set; Basic Medical Biochemistry, 6th Ed.)
4. Regulation of Aldosterone Secretion
Three main stimuli control aldosterone release:
4.1 Angiotensin II (Ang II) - Primary Regulator
The renin-angiotensin-aldosterone system (RAAS) is the dominant regulatory pathway:
- Reduced renal perfusion pressure → juxtaglomerular cells release renin
- Renin cleaves angiotensinogen (from liver) → Angiotensin I
- ACE (in lung) converts Ang I → Angiotensin II
- Ang II binds AT1 receptors on zona glomerulosa cells → activates phospholipase C → generates IP₃ and DAG → elevates intracellular Ca²⁺ → stimulates CYP11B2 and StAR → aldosterone secretion
Ang II also separately stimulates Ca²⁺ influx through voltage-sensitive Ca²⁺ channels, which sustains the secretory response.
4.2 Hyperkalemia - Second Most Important Stimulus
Elevated plasma K⁺ directly depolarizes zona glomerulosa cell membranes → opens voltage-sensitive Ca²⁺ channels → raises intracellular Ca²⁺ → stimulates aldosterone synthesis. This creates a direct feedback loop: aldosterone causes renal K⁺ excretion → lowers plasma K⁺ → reduces the stimulus.
4.3 ACTH - Minor Stimulus
ACTH (from the anterior pituitary) stimulates aldosterone acutely by increasing StAR activity and cholesterol availability, but it is a much weaker and less sustained stimulus than Ang II or K⁺. It does not account for long-term aldosterone regulation.
4.4 Inhibitors of Aldosterone Secretion
- Atrial natriuretic peptide (ANP) - released from the heart in response to volume expansion, directly inhibits aldosterone secretion
- Somatostatin - inhibitory
- High sodium intake - suppresses renin → less Ang II → less aldosterone
(Brenner and Rector's The Kidney; Henry's Clinical Diagnosis and Management by Laboratory Methods)
5. Mechanism of Action
Aldosterone acts through both genomic (classical) and non-genomic pathways.
5.1 Genomic Pathway (Primary)
This is the main mechanism for aldosterone's effects on epithelial transport:
- Aldosterone diffuses across the cell membrane (being lipid-soluble)
- It binds to the mineralocorticoid receptor (MR) in the cytoplasm
- The hormone-receptor complex translocates to the nucleus
- The complex binds to specific hormone response elements (HREs) on DNA
- It alters transcription of target genes
Key genomic effects in the kidney (principal cells of the distal nephron/collecting duct):
- Early response (30-60 min): Induction of SGK1 (serum- and glucocorticoid-regulated kinase 1) - a serine-threonine kinase that phosphorylates Nedd4-2 and prevents it from internalizing ENaC → more ENaC channels remain in the apical membrane → increased Na⁺ reabsorption
- Late response (hours): Increased transcription of all three ENaC subunits (α, β, γ) → more channel protein synthesized
- Basolateral effects: Increased expression of Na⁺-K⁺-ATPase α₁ and β₁ subunits → enhanced Na⁺ pumping from cell into blood
- Net result: Na⁺ is reabsorbed, water follows osmotically → volume expansion; K⁺ and H⁺ are secreted in exchange
The aldosterone-sensitive distal nephron (ASDN) includes the distal convoluted tubule (DCT), connecting tubule (CNT), and collecting duct - all rich in MR.
(Ganong's Review of Medical Physiology, 26th Ed.; Brenner and Rector's The Kidney)
5.2 Non-Genomic Pathway
Evidence shows aldosterone can also bind to the cell membrane and, via IP₃ as a second messenger, rapidly activate Na⁺-K⁺ exchangers independent of gene transcription. However, the principal physiological effects are genomically mediated.
5.3 Why Doesn't Cortisol Activate the MR?
The MR has similar affinity for both cortisol and aldosterone. However, cortisol does NOT normally occupy renal MRs because of the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), expressed in aldosterone-target tissues. This enzyme rapidly converts cortisol to the inactive cortisone, preventing it from binding MR. If 11β-HSD2 is inhibited (e.g., by excessive liquorice/glycyrrhizin) or congenitally absent, cortisol floods the MR and causes the syndrome of Apparent Mineralocorticoid Excess (AME) - presenting like hyperaldosteronism with low renin and low aldosterone.
(Ganong's Review of Medical Physiology, 26th Ed.)
6. Physiological Effects
6.1 Kidney
- Na⁺ retention in the collecting duct and distal tubule → water retention → expansion of ECF volume
- K⁺ excretion into urine - the most clinically important countertransport effect
- H⁺ secretion → mild metabolic alkalosis
- Cl⁻ reabsorption - secondary to sodium
6.2 Colon
Aldosterone enhances Na⁺ absorption and K⁺ secretion in the colonic epithelium - these effects become clinically relevant in patients with renal failure.
6.3 Salivary Glands and Sweat Glands
Reduces Na⁺ concentration in saliva and sweat, conserving sodium.
6.4 Cardiovascular Effects (Non-Epithelial)
A 2024 review in the Journal of the American Heart Association (PMID 38497438) emphasizes that aldosterone's adverse effects extend well beyond blood pressure:
- Cardiac fibrosis - MR activation in cardiac fibroblasts promotes collagen deposition
- Left ventricular hypertrophy (LVH)
- Vascular inflammation and stiffness
- Endothelial dysfunction
- Renal fibrosis and proteinuria - through non-hemodynamic mechanisms
- Sympathetic nervous system activation - raising cardiovascular risk
A 2025 review in International Journal of Molecular Sciences (PMID 39859256) catalogs non-hypertensive effects including effects on adipose tissue, brain, bone, and immune function.
7. Clinical Disorders
7.1 Primary Hyperaldosteronism (Conn Syndrome)
Definition: Autonomous, renin-independent hypersecretion of aldosterone from the adrenal gland.
Causes (three main):
| Cause | Frequency | Notes |
|---|---|---|
| Bilateral idiopathic hyperplasia | ~60% | Older patients, less severe hypertension; medical management preferred |
| Aldosterone-producing adenoma (Conn syndrome) | ~35% | Solitary, usually <2 cm; female > male (2:1); 30-40s onset; surgical cure possible |
| Familial hyperaldosteronism (FH-I to FH-IV) | ~5% | FH-I = glucocorticoid-remediable aldosteronism; CYP11B2 under ACTH-responsive CYP11B1 promoter |
Molecular genetics of aldosterone-producing adenomas:
- ~50% carry somatic KCNJ5 mutations (encodes GIRK4 potassium channel) → loss of K⁺ selectivity → Na⁺ influx → depolarization → Ca²⁺ entry → chronic activation of aldosterone synthase
- Other mutations: ATP1A1, ATP2A3, CACNA1D, CTNNB1
Clinical features:
- Hypertension (often treatment-resistant) - most common presentation
- Hypokalemia (not invariable): muscle weakness, fatigue, paresthesias, polyuria, polydipsia
- Metabolic alkalosis (due to H⁺ loss)
- Low plasma renin activity (PRA) - hallmark of primary disease
- Increased risk of stroke and MI beyond what blood pressure alone predicts
Diagnosis:
- Screening: aldosterone-to-renin ratio (ARR) - elevated in primary hyperaldosteronism
- Confirmatory: aldosterone suppression test (saline infusion or oral salt loading)
- Subtype differentiation: adrenal CT, and adrenal vein sampling (gold standard)
Treatment:
- Adrenal adenoma (Conn syndrome): adrenalectomy (laparoscopic) - curative in most
- Bilateral hyperplasia: aldosterone antagonists (spironolactone or eplerenone); potassium-sparing diuretics
- FH-I: low-dose glucocorticoid (suppresses ACTH → suppresses chimeric gene → reduces aldosterone)
(Robbins, Cotran & Kumar Pathologic Basis of Disease)
7.2 Secondary Hyperaldosteronism
Definition: Elevated aldosterone in response to appropriate (physiological) activation of the RAAS - plasma renin is HIGH (distinguishing it from primary).
Causes:
- Decreased renal perfusion: renal artery stenosis, arteriolar nephrosclerosis
- Effective arterial hypovolemia / edema states: congestive heart failure, cirrhosis, nephrotic syndrome
- Pregnancy: estrogen increases renin substrate (angiotensinogen)
Treatment: Directed at the underlying cause.
7.3 Hypoaldosteronism
Causes:
- Primary adrenal insufficiency (Addison's disease) - loss of all adrenal cortical zones
- Hyporeninemic hypoaldosteronism - most common cause; reduced renin release from the kidney (often in diabetic nephropathy, chronic interstitial nephritis); presents with hyperkalemia and mild acidosis without overt adrenal failure
- Congenital aldosterone synthesis defects
- Pseudohypoaldosteronism - normal aldosterone but end-organ resistance to it (MR mutation)
Clinical features: Hyperkalemia, hyponatremia, hypotension, metabolic acidosis
Treatment: Fludrocortisone (synthetic mineralocorticoid replacement)
8. Laboratory Assessment
| Test | Normal Value | Interpretation |
|---|---|---|
| Plasma aldosterone | 4-21 μg/dL (upright) | Elevated in primary/secondary hyperaldosteronism |
| Plasma renin activity (PRA) | Variable | Low in primary; high in secondary |
| Aldosterone-to-renin ratio (ARR) | <30 (ng/dL)/(ng/mL/hr) | >30-40 suggests primary hyperaldosteronism |
| Urinary aldosterone (24h) | 5-19 μg/day | Elevated if secretion is autonomous |
| Serum K⁺ | 3.5-5.0 mEq/L | Hypokalemia in severe hyperaldosteronism |
The unique metabolism of aldosterone produces an "acid-labile conjugate" (18-glucuronide) that, unlike other steroid metabolites, is hydrolyzed back to free aldosterone at pH 1.0 - this forms the basis of classic urine assay methodology.
(Henry's Clinical Diagnosis and Management by Laboratory Methods)
9. Pharmacology - Aldosterone Antagonists (MR Antagonists)
| Drug | Type | Selectivity | Key Uses |
|---|---|---|---|
| Spironolactone | Steroidal MRA | Non-selective (also blocks androgen/progesterone receptors) | Heart failure (HFrEF), primary hyperaldosteronism, resistant hypertension, ascites |
| Eplerenone | Steroidal MRA | Selective for MR | Heart failure, hypertension; fewer sex hormone side effects |
| Finerenone | Non-steroidal MRA | Highly selective MR | CKD with type 2 diabetes (FDA approved 2021); reduces renal and cardiovascular events |
Mechanism: All competitively block the MR → reduce aldosterone-induced Na⁺ reabsorption → potassium-sparing diuresis
Key clinical trial evidence: Both spironolactone (RALES trial) and eplerenone (EPHESUS trial) reduced mortality in heart failure.
Side effects of spironolactone: Gynecomastia and breast pain (~10% of men) from anti-androgenic effects - switch to eplerenone if intolerable.
(Goodman & Gilman's Pharmacological Basis of Therapeutics; Katzung's Basic and Clinical Pharmacology, 16th Ed.)
10. The Renin-Angiotensin-Aldosterone System (RAAS) - Overview
Reduced renal perfusion / Na⁺ depletion / β-adrenergic stimulation
↓
Renin (from JGA cells)
↓
Angiotensinogen → Angiotensin I
↓ (ACE, in lung)
Angiotensin II
↙ ↘
Vasoconstriction Aldosterone secretion
(AT1 receptor) (adrenal glomerulosa)
↓
Na⁺ and H₂O retention, K⁺ excretion
↓
Restored blood pressure/volume
↓
Renin secretion suppressed
(negative feedback)
The RAAS is the most important integrated axis controlling blood pressure, fluid balance, and electrolytes. Drugs targeting this axis (ACEi, ARBs, MRAs) are cornerstones of cardiovascular and renal medicine.
11. Familial Hyperaldosteronism - Genetic Forms
| Type | Gene Defect | Mechanism | Treatment |
|---|---|---|---|
| FH-I (glucocorticoid-remediable aldosteronism) | CYP11B1/CYP11B2 chimeric gene on chr 8 | ACTH drives aldosterone production | Low-dose glucocorticoid (dexamethasone) |
| FH-II | CLCN2 (chloride channel) | Unknown mechanism | Spironolactone/surgery |
| FH-III | KCNJ5 germline mutation | Depolarization of glomerulosa cells | Surgery (marked bilateral hyperplasia) |
| FH-IV | CACNA1H | Calcium channel gain-of-function | Variable |
12. Summary Table
| Feature | Detail |
|---|---|
| Hormone class | Steroid (mineralocorticoid) |
| Source | Zona glomerulosa, adrenal cortex |
| Chemical signature | C18 aldehyde group; no 17α-OH |
| Key biosynthetic enzyme | Aldosterone synthase (CYP11B2) |
| Daily secretion | 50-200 μg/day |
| Plasma half-life | ~20 min |
| Primary stimuli | Ang II > K⁺ > ACTH |
| Primary inhibitor | ANP, somatostatin |
| Receptor | Mineralocorticoid receptor (MR), nuclear |
| Key genomic targets | ENaC (α, β, γ subunits), SGK1, Na⁺-K⁺-ATPase |
| Net renal effect | Na⁺ and H₂O retention; K⁺ and H⁺ excretion |
| Deficiency | Addison's disease, hyporeninemic hypoaldosteronism |
| Excess | Primary (Conn) or secondary hyperaldosteronism |
| Pharmacological block | Spironolactone, eplerenone, finerenone |
13. Conclusion
Aldosterone is far more than a simple "salt-retaining hormone." It is a master regulator of sodium and potassium homeostasis, blood pressure, and extracellular fluid volume, acting primarily through genomic regulation of ENaC and Na⁺-K⁺-ATPase in the distal nephron. Its secretion is tightly controlled through the RAAS and plasma potassium via calcium-dependent signaling in the zona glomerulosa. When produced in excess - whether from an autonomous adenoma (Conn syndrome), bilateral hyperplasia, or chronic RAAS activation - it causes hypertension, hypokalemia, and end-organ damage extending well beyond blood pressure control. Conversely, its deficiency threatens life through hyperkalemia and hypotension. Mineralocorticoid receptor antagonists, from the classic spironolactone to the newer nonsteroidal finerenone, harness our understanding of aldosterone physiology to treat heart failure, hypertension, CKD, and primary hyperaldosteronism.
Sources:
- Brenner and Rector's The Kidney, 2-Volume Set - Chapters 6, 12
- Ganong's Review of Medical Physiology, 26th Edition - Chapter 19
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 24
- Goodman & Gilman's Pharmacological Basis of Therapeutics
- Katzung's Basic and Clinical Pharmacology, 16th Edition
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Basic Medical Biochemistry, 6th Edition
- Brown JM. Adverse Effects of Aldosterone: Beyond Blood Pressure. J Am Heart Assoc. 2024. [PMID: 38497438]
- Ekman N et al. Non-Hypertensive Effects of Aldosterone. Int J Mol Sci. 2025. [PMID: 39859256]
This is a shared conversation. Sign in to Orris to start your own chat.